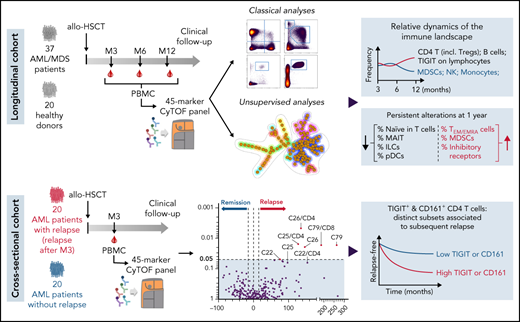
Key Points
Immunoregulatory cell compartments show distinct dynamics and substantial persistent alterations at 1 year after allo-HSCT.
High levels of TIGIT and CD161 expression on CD4 T cells early after allo-HSCT are associated with subsequent relapse in patients with AML.

Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the most effective treatment for selected patients with acute myeloid leukemia (AML) and relies on a “graft-versus-leukemia” effect (GVL) where donor T lymphocytes mediate control of malignant cell growth. However, relapse remains the major cause of death after allo-HSCT. In various malignancies, several immunoregulatory mechanisms have been shown to restrain antitumor immunity, including ligand-mediated engagement of inhibitory receptors (IRs) on effector cells, and induction of immunosuppressive cell subsets, such as regulatory T cells (Tregs) or myeloid-derived suppressor cells (MDSCs). Relapse after HSCT remains a major therapeutic challenge, but immunoregulatory mechanisms involved in restraining the GVL effect must be better deciphered in humans. We used mass cytometry to comprehensively characterize circulating leukocytes in 2 cohorts of patients after allo-HSCT. We first longitudinally assessed various immunoregulatory parameters highlighting specific trends, such as opposite dynamics between MDSCs and Tregs. More generally, the immune landscape was stable from months 3 to 6, whereas many variations occurred from months 6 to 12 after HSCT. Comparison with healthy individuals revealed that profound alterations in the immune equilibrium persisted 1 year after HSCT. Importantly, we found that high levels of TIGIT and CD161 expression on CD4 T cells at month 3 after HSCT were distinct features significantly associated with subsequent AML relapse in a second cross-sectional cohort. Altogether, these data provide global insights into the reconstitution of the immunoregulatory landscape after HSCT and highlight non-canonical IRs associated with relapse, which could open the path to new prognostic tools or therapeutic targets to restore subverted anti-AML immunity.
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the most effective consolidation treatment for patients with intermediate- to high-risk acute myeloid leukemia (AML). The efficacy of allo-HSCTmostly relies on the ability of donor T cells to eliminate tumor cells, a process referred to as the graft-versus-leukemia (GVL) effect.1-3 However, relapse still occurs in ∼40% of patients and remains the main cause of mortality after allo-HSCT.4-6 Reduced antigen presentation via HLA class II and overexpression of ligands for inhibitory receptors (IRs) have been shown recently on relapsing AML blasts, with concomitant upregulation of IRs on T cells.7-9 However, although research efforts have largely focused on graft-versus-host disease (GVHD), the immunological mechanisms behind the effectiveness of, or escape from, GVL responses still need closer scrutiny in humans.3,10
Immunoregulatory mechanisms hindering efficient antitumor immune responses mainly fall into 2 categories: (1) cell-intrinsic, via ligand-mediated engagement of IRs expressed on effector cells (eg, PD-1, CTLA-4, TIGIT, Tim-3, Lag-3, and BTLA), and (2) cell-extrinsic, via immunosuppressive cell subsets, such as regulatory T cells (Tregs) or myeloid-derived suppressor cells (MDSCs).11-13 In contrast to Tregs, a well-defined cell type described in health and diseases, MDSCs represent a more heterogeneous population (monocytic or granulocytic) known to play a major role in restraining antitumor T-cell responses,12 but also possibly in limiting GVHD.14 Other cell subsets, such as type 2 innate lymphoid cells (ILC2s), may also exert regulatory functions in cancer contexts.15 Indeed, we identified a protumor ILC2-MDSC axis in humans16,17 that was also associated with tolerance induction in a mouse model of GVHD.18
The process of immune reconstitution after allo-HSCT is slow, complex, and yet incompletely understood (reviewed in Ogonek et al19 and Velardi et al20). In contrast to early recovery of innate cells (mostly neutrophils and natural killer [NK] cells), the reconstitution of the adaptative system is much slower and is influenced by multiple patient-, disease-, and transplant-related cofactors. Main transplant and posttransplant cofactors influencing T- and B-cell reconstitution include recipients’ age, graft source, the use of T-cell depleting agents, and the presence of GVHD.20,21 Immune reconstitution of all main immune cell subsets had been studied individually for many years by flow cytometry, but systems-level analyses by high-dimensional, single-cell technologies such as mass cytometry provide a wider and less biased picture of the overall process of innate and adaptative immune reconstitution.22-24
In this work, our aims were twofold: (1) to study, globally and longitudinally, the immunoregulatory landscape and (2) to identify whether and which immune cell subsets or immunoregulatory features could be associated with the emergence of subsequent relapse after allo-HSCT. As immune regulatory mechanisms involve a complex and interconnected network of diverse immunosuppressive cells subsets and IRs,25 mass cytometry was used to study circulating leukocytes after allo-HSCT in (1) a longitudinal cohort up to 1 year after HSCT and (2) a cross-sectional cohort at month 3 after transplant, including 2 groups matched for most relevant clinical variables, with patients showing either subsequent relapse or persistent long-term remission.
Methods
Human biological samples
The 2 cohorts of HSCT recipients are detailed in supplemental Methods (available on the Blood Web site). The first cohort included patients with AML or myelodysplastic syndrome (MDS), with prespecified sampling at 3, 6, and 12 months after HSCT in the absence of hematological relapse,26,27 and enabled us to analyze the global longitudinal changes in the immunoregulatory landscape. The second cohort included patients with AML, from which we retrospectively selected samples at 3 months after HSCT from 20 patients who subsequently relapsed and 20 patients without relapse, matched for age at transplantation, donor type, and AML cytogenetic and molecular risk.28 Ethics approval was obtained according to French regulation (detailed in supplemental Methods). Blood specimens were also collected from healthy volunteers (median 36 years, interquartile range [IQR], 29-51) through the French Blood Bank.
Mass cytometry panel design and staining
We designed a 45-marker panel (including 44 metal-labeled antibodies and cisplatin as viability stain) as reported in supplemental Table 1. The protocol is detailed in the supplemental Methods.
Mass cytometry data analyses and statistics
Cell events were acquired on the CyTOF Helios Mass Cytometer (Fluidigm), and data were normalized as detailed in the supplemental Methods. Data were analyzed by classic gating and by using the unsupervised clustering algorithms FlowSOM and Uniform Manifold Approximation and Projection (UMAP) for dimensional reduction.29,30 The analyses were performed using FlowJo (v10.6), the Cytobank platform (Beckman Coulter), and R (v4.0.2), as detailed in supplemental Methods. Nonparametric (Mann-Whitney, Wilcoxon, and log-rank) tests were used as detailed in the supplemental Methods.
Results
Immunoregulatory landscape dynamics following allo-HSCT
We first sought to comprehensively analyze longitudinal changes in the abundance and functional phenotype of peripheral immune cell subsets after allo-HSCT. We included 37 patients with AML or MDS from the first longitudinal cohort, in whom peripheral blood mononuclear cells (PBMCs) were collected at months 3 (M3), M6, and M12 after transplantation, as well as 20 healthy donors (Figure 1A). Patients’ characteristics are described in Table 1 and supplemental Table 2. Twelve patients had relapse at a later time after sampling (particularly late, with a median of 15 [IQR, 8-69] months after HSCT). Most patients with AML had intermediate or adverse factors according to the European LeukemiaNet classification. Relapsed patients were significantly more likely to have undergone reduced-intensity conditioning and were not in complete remission at the time of transplantation. All other characteristics were similar between the groups who did or did not relapse. As expected, patients with relapsed disease had worse survival (Table 1).
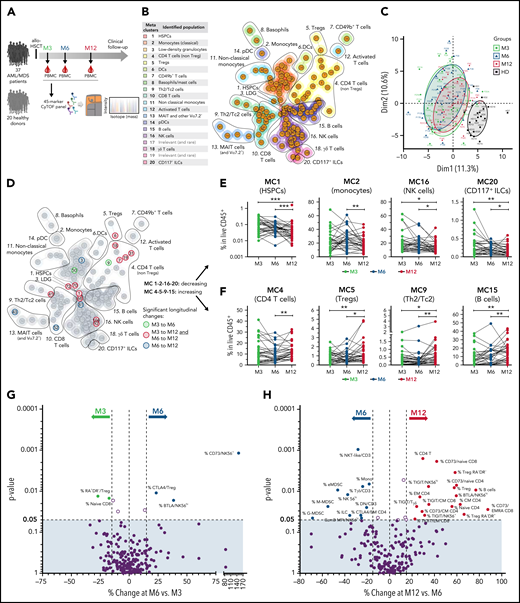
Longitudinal changes in the immunoregulatory landscape after allo-HSCT. (A) Thirty-seven patients with AML/MDS were included in the longitudinal cohort. PBMCs were collected at months 3, 6, and 12 after transplantation and analyzed by mass cytometry. Twenty healthy donors were also analyzed. (B) Unsupervised cell-clustering analysis by FlowSOM, with 18 relevant metaclusters. (C) Principal component analysis (using FlowSOM data) of all tested samples from patients at the indicated time points (M3, n = 34; M6, n = 31; M12, n = 33) and from healthy donors. (D) Highlight of cell clusters showing significant longitudinal variations between the indicated time points. (E-F) Frequencies among PBMCs of cell metaclusters decreasing (E) or increasing (F) over time. (G-H) Volcano plots showing changes between M3 and M6 (G) and between M6 and M12 (H) in immune parameters assessed by classic manual gating. Significant parameters with percentage change >15% are annotated (percentage of cell population refers to total CD45+ live cells or to a parent cell subset when indicated by “/subset”). P values by Wilcoxon signed-rank tests. *P < .05; **P < .01; ***P < .001.
Longitudinal changes in the immunoregulatory landscape after allo-HSCT. (A) Thirty-seven patients with AML/MDS were included in the longitudinal cohort. PBMCs were collected at months 3, 6, and 12 after transplantation and analyzed by mass cytometry. Twenty healthy donors were also analyzed. (B) Unsupervised cell-clustering analysis by FlowSOM, with 18 relevant metaclusters. (C) Principal component analysis (using FlowSOM data) of all tested samples from patients at the indicated time points (M3, n = 34; M6, n = 31; M12, n = 33) and from healthy donors. (D) Highlight of cell clusters showing significant longitudinal variations between the indicated time points. (E-F) Frequencies among PBMCs of cell metaclusters decreasing (E) or increasing (F) over time. (G-H) Volcano plots showing changes between M3 and M6 (G) and between M6 and M12 (H) in immune parameters assessed by classic manual gating. Significant parameters with percentage change >15% are annotated (percentage of cell population refers to total CD45+ live cells or to a parent cell subset when indicated by “/subset”). P values by Wilcoxon signed-rank tests. *P < .05; **P < .01; ***P < .001.
We designed a mass-cytometry panel to assess the distribution of all main immune cell types, with a particular focus on immunoregulatory cells and receptors (ie, Tregs, MDSCs, and a range of IRs). Unsupervised clustering analysis using FlowSOM (Figure 1B; supplemental Figure 1A-B) enabled identification of 121 cell clusters classified into 18 relevant metaclusters (MCs) of various relative abundances (supplemental Figure 1A). Principal component analysis revealed very distinct immune profiles in patients, compared with healthy donors, whereas time point after allo-HSCT did not segregate the patients’ samples (Figure 1C).
We next tested variations of each cell cluster in paired samples at the different time points and found very few changes between M3 and M6, whereas the frequencies of several cell clusters significantly varied from M6 to M12 (Figure 1D; detailed in supplemental Figure 1C). At the MC level, we found that the proportions of MC1 (hematopoietic stem/progenitor cells [HSPCs]), MC2 (monocytes), MC16 (NK cells), and MC20 (CD117+ ILCs) decreased (Figure 1E), whereas those of MC4 (CD4 T cells), MC5 (Tregs), MC9 (Th2/Tc2 cells), and MC15 (B cells) increased over time (Figure 1F). Analyses using classic manual gating strategies (supplemental Figure 2) confirmed that only a few significant changes occurred between M3 and M6 (Figure 1G), whereas several immune parameters varied between M6 and M12 (Figure 1H). Regarding immunoregulatory cell populations, all 3 MDSC subsets (monocytic, granulocytic, and early-stage MDSCs31) were elevated in patients at M3, as compared with healthy donors (supplemental Figure 3). Of note, the frequency of e-MDSC and M-MDSCs subsets decreased from M6 to M12 (Figure 1H). In contrast, Treg frequencies among PBMCs increased, as a reflection of increased total CD4 T cells. More specifically, only memory (CD45RA−), both “activated” (HLA-DR+) and “cytokine-secreting” (HLA-DR−),32,33 but not naive or resting Tregs were significantly increased at M12 (Figure 1H). Besides, the proportion of cells expressing the TIGIT IR significantly increased from M6 to M12, not only among CD8 TCM cells, but also among γδ-T and NK cells (Figure 1H). There was also an increase in BTLA positivity on T-cell subsets at M12 when compared with M3 (supplemental Figure 4). Expression of the other IRs (PD-1, LAG-3, Tim-3, and CTLA-4) did not change significantly over time.
Although limited by the number of patients, results of subgroup analyses including clinical parameters suggest an impact of ATG serotherapy in the longitudinal study of the CD4 T-cell compartment (lower frequencies of CD4 T cells up to M12 in patients with ATG infusion; supplemental Figure 5) and of cytomegalovirus reactivation on CD8 T- and B-cell relative abundance (supplemental Figure 6).
Persistent alterations at 1 year after allo-HSCT
We then investigated whether and how the circulating immunoregulatory landscape remained altered 1 year after transplantation, when compared with the immune equilibrium at homeostasis. Hence, we first identified cell metaclusters with significantly different relative abundance between patients at M12 and healthy donors (Figure 2A-B). This analysis showed reduced relative levels of MC1 (HSPCs), MC4 (CD4 T cells), MC6 (DCs), MC13 (MAIT and other TCR Vα7.2+ cells), MC14 (pDCs), and MC20 (CD117+ ILCs) and elevated relative levels of MC16 (NK cells) in patients compared with healthy donors. We then generated a global atlas of immune subsets found to be reduced (Figure 2C, left) or elevated (Figure 2C, right) in patients at M12, emphasizing the profound and diverse persistent alterations in the immune equilibrium in transplant recipients. These analyses confirmed long-term delayed relative reconstitution of CD4 T cells, MAIT cells, pDCs, non-classical monocytes, and the 3 subsets of innate lymphoid cells (ILC1, ILC2, and ILC3/P), in contrast to the immature (CD56brightCD16−) NK cells that were overrepresented in patients. Moreover, the T-cell compartment was biased toward the effector memory and terminally differentiated phenotype, mirrored by a persistent lower frequency of naive and central memory T cells. Regarding immunoregulatory cell populations, MDSCs only partly normalized with persistently elevated relative levels of cells with phenotypes of e-MDSCs and G-MDSCs, whereas M-MDSC levels were similar to those of healthy donors. Total Treg frequency among CD4 T cells was also similar in patients and healthy donors, but the Treg compartment was biased toward activated (CD45RA−HLA-DR+) Tregs. Importantly, most IRs (TIGIT, PD-1, Tim-3, LAG-3, and CTLA-4) were expressed on a higher proportion of CD8 and/or CD4 T cells and NK cells from the patients (Figure 2D). Surprisingly, the opposite was observed for the BTLA IR, which was expressed at lower frequencies on T cells from patients. This may have reflected the deficiency in naive T cells in patients, as BTLA downregulation is a known feature of T-cell differentiation.34 Of note, the intracellular cytotoxic molecule granzyme B was expressed at higher levels in most T-cell subsets, and in NK cells.

Persistent alterations in the immunoregulatory landscape at M12 after allo-HSCT. (A-B) Frequencies of cell metaclusters (FlowSOM) among PBMCs from patients at M12 after transplantation were compared with those of healthy donors. Six of 18 cell MCs were significantly decreased (A) and 1 was increased (B). Mann-Whitney tests: *P < .05; **P < .01; ***P < .001; ****P < .0001. (C) Each immune cell subset (percentage of cell subset among total CD45+ live cells or among a parent cell subset when indicated by "/subset") assessed by classic manual gating was also compared between patients at M12 and healthy donors. Significant (Mann-Whitney test) parameters are annotated. (D) Altered expression of immunoregulatory molecules in indicated T- and NK-cell subsets. Circles indicate significantly increased (red) or decreased (gray) median expression of the regulatory molecules in patients at M12 after HSCT, compared with healthy donors.
Persistent alterations in the immunoregulatory landscape at M12 after allo-HSCT. (A-B) Frequencies of cell metaclusters (FlowSOM) among PBMCs from patients at M12 after transplantation were compared with those of healthy donors. Six of 18 cell MCs were significantly decreased (A) and 1 was increased (B). Mann-Whitney tests: *P < .05; **P < .01; ***P < .001; ****P < .0001. (C) Each immune cell subset (percentage of cell subset among total CD45+ live cells or among a parent cell subset when indicated by "/subset") assessed by classic manual gating was also compared between patients at M12 and healthy donors. Significant (Mann-Whitney test) parameters are annotated. (D) Altered expression of immunoregulatory molecules in indicated T- and NK-cell subsets. Circles indicate significantly increased (red) or decreased (gray) median expression of the regulatory molecules in patients at M12 after HSCT, compared with healthy donors.
Identification of an immune profile associated with subsequent late relapse
In this longitudinal cohort, late AML/MDS relapse occurred in 12 patients (median, 15 months after HSCT). We investigated whether the immunoregulatory profile showed particular features in these patients with late relapse compared with those in long-term persistent remission (median follow-up, 9.8 [IQR 8.3-9.9] years). When assessed at M3, frequencies of naive/CM CD8 T cells and γδ T cells were at higher levels in patients with persistent remission (with only 9 patients of the relapsing group available at this time point; supplemental Figure 7A). We then compared immune parameters at the M12 (or last available) time point, and found a slight segregation between patients with or without subsequent tumor relapse in a principal component analysis (Figure 3A). Most cell metaclusters were found at similar relative levels between both groups (supplemental Figure 7B). However, frequencies of MC18 (γδ T cells) and MC13 (MAIT cells) were significantly decreased in patients with subsequent relapse, compared with those with sustained remission (Figure 3B). Among all single immune parameters differing between both patient groups (Figure 3C), we identified that increased proportions of all innate-like T-cell subsets (MAIT, γδ T, and NKT cells) and of naive CD8 T cells were associated with long-term remission. Notably, frequencies of TIGIT-expressing cells were increased among various T-cell subsets in patients with subsequent relapse (Figure 3C).
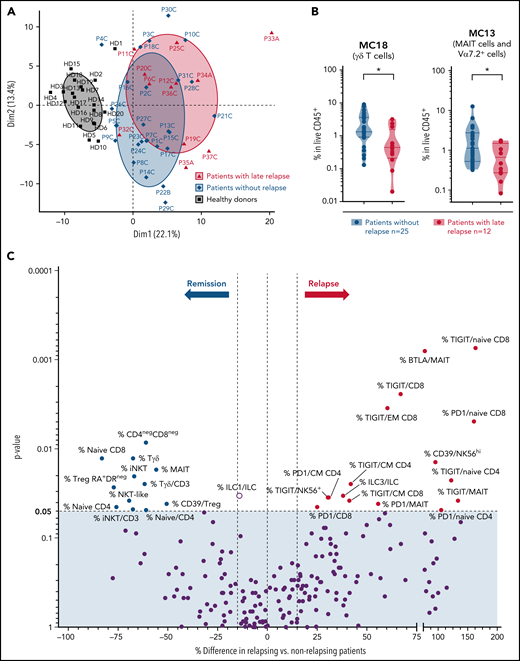
Immune parameters associated with late relapse in the longitudinal cohort. (A) Principal component analysis (using FlowSOM data) of last available samples from patients with (n = 12) or without (n = 25) subsequent tumor relapse, as well as of healthy donors. (B) Frequencies of cell MCs (FlowSOM) in last available samples were compared between patients with or without subsequent tumor relapse. Two of 18 cell MCs were significantly decreased in patients with subsequent relapse. These 2 cell MCs are shown; other MCs (similar in both groups) are shown in supplemental Figure 3B. *P < .05, by Mann-Whitney test. (C) Volcano plots showing differences in immune parameters at the last available time point between patients with or without relapse, assessed by classic manual gating. P-values by Mann-Whitney test. Significantly different parameters between both groups are annotated (percentage cell population is among total CD45+ live cells or among a parent cell subset, when indicated by “/subset”).
Immune parameters associated with late relapse in the longitudinal cohort. (A) Principal component analysis (using FlowSOM data) of last available samples from patients with (n = 12) or without (n = 25) subsequent tumor relapse, as well as of healthy donors. (B) Frequencies of cell MCs (FlowSOM) in last available samples were compared between patients with or without subsequent tumor relapse. Two of 18 cell MCs were significantly decreased in patients with subsequent relapse. These 2 cell MCs are shown; other MCs (similar in both groups) are shown in supplemental Figure 3B. *P < .05, by Mann-Whitney test. (C) Volcano plots showing differences in immune parameters at the last available time point between patients with or without relapse, assessed by classic manual gating. P-values by Mann-Whitney test. Significantly different parameters between both groups are annotated (percentage cell population is among total CD45+ live cells or among a parent cell subset, when indicated by “/subset”).
Cross-sectional cohort study reveals T-cell subsets associated with subsequent AML relapse
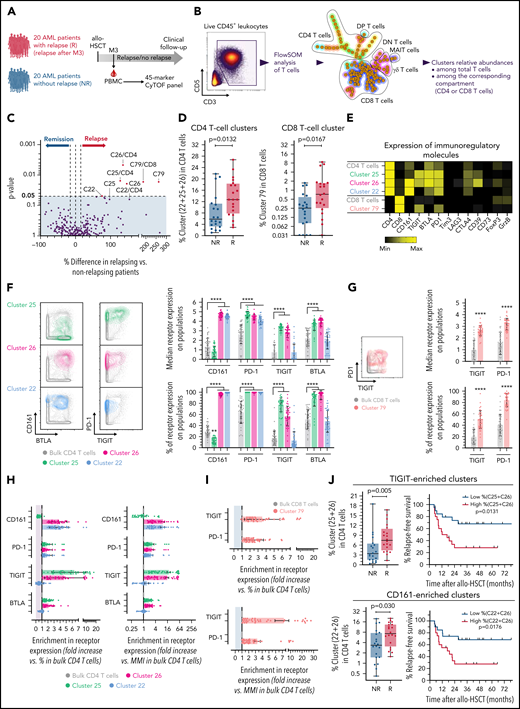
The first cohort had substantial limitations to studying immune parameters associated with relapse (few and only late relapses, disease heterogeneity, no matching for confounding risk factors). We thus next investigated a homogenous retrospective cohort including 40 patients at M3 after transplant (Figure 4A). Patients’ characteristics are described in Table 2 and supplemental Table 2. Twenty patients with AML relapse (after M3, median, 8.7 months) and 20 patients without documented relapse (median follow-up, 32 [IQR 17-52] months) were matched according to known disease relapse risks, as described in “Methods.” All other parameters were similar between both groups, with only nonsignificant higher risk of subsequent chronic GVHD, and, as expected, worse survival in relapsing patients (Table 2). We first performed a FlowSOM analysis of all PBMCs from all patients and included 28 subset-defining phenotypic markers (supplemental Figure 8). In this well-matched cohort, we found no difference between both patient groups in the frequency of any FlowSOM-generated cell clusters (data not shown). We next focused on the T-cell compartment and performed an ad hoc FlowSOM analysis of pregated total T cells (Figure 4B), with all functional markers included for the cluster generation (supplemental Table 1). Although none of the clusters was enriched in nonrelapsing patients, 3 CD4 T-cell clusters (C25, C26, C22) and 1 rare CD8 T-cell cluster (C79) were found at increased frequencies in patients with subsequent relapse (Figure 4C-D). In comparison with bulk CD4 or CD8 T cells, all of these cell clusters expressed higher surface levels of several IRs, and higher CD39 expression (for C25 and C26; Figure 4E).
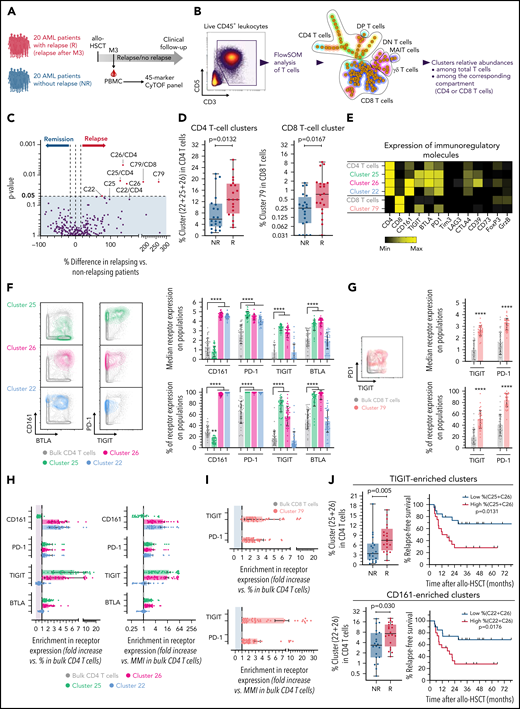
Inhibitory receptors expression on T cells is associated with subsequent AML tumor relapse in the cross-sectional cohort. (A) PBMCs from 40 patients with AML, with (n = 20) or without (n = 20) documented subsequent tumor relapse, were collected at M3 after transplantation and analyzed by mass cytometry. (B) Unsupervised cell clustering of the T-cell compartment was performed with FlowSOM. (C) Volcano plots showing differences between patients with or without relapse in frequencies of FlowSOM-generated T-cell clusters. P-values by Mann-Whitney test. Clusters that were significantly different between both groups are annotated (percentage of cell clusters is among total CD3+ T cells or among CD8 or CD4 T cells, when indicated). (D) Frequencies of identified cell clusters among CD4 or CD8 T cells in relapsing (R) and nonrelapsing (NR) patients. (E) Heat map showing expression levels of indicated immunoregulatory molecules in relapse-associated cell clusters. (F) Expression of CD161, PD-1, TIGIT, and BTLA on total and relapse-associated CD4 T-cell clusters. (G) Expression of PD-1 and TIGIT on total and on the relapse-associated CD8 T-cell cluster. (H-I) Enrichment in the indicated IR positivity or expression density (median metal intensity, MMI) in relapse-associated clusters relative to total CD4 (H) and CD8 (I) T cells. (J) Frequencies of combined TIGIT clusters (C25+C26) or CD161 clusters (C22+C26) in relapsing (R) and nonrelapsing (NR) patients. Relapse-free survival (RFS) assessed using the Kaplan-Meier approach in patients with high (>median) vs low (<median) frequencies of indicated cell clusters are shown (right). P values by Mann-Whitney and log-rank tests, to compare cluster frequency and RFS, respectively. ****P < .0001.
Inhibitory receptors expression on T cells is associated with subsequent AML tumor relapse in the cross-sectional cohort. (A) PBMCs from 40 patients with AML, with (n = 20) or without (n = 20) documented subsequent tumor relapse, were collected at M3 after transplantation and analyzed by mass cytometry. (B) Unsupervised cell clustering of the T-cell compartment was performed with FlowSOM. (C) Volcano plots showing differences between patients with or without relapse in frequencies of FlowSOM-generated T-cell clusters. P-values by Mann-Whitney test. Clusters that were significantly different between both groups are annotated (percentage of cell clusters is among total CD3+ T cells or among CD8 or CD4 T cells, when indicated). (D) Frequencies of identified cell clusters among CD4 or CD8 T cells in relapsing (R) and nonrelapsing (NR) patients. (E) Heat map showing expression levels of indicated immunoregulatory molecules in relapse-associated cell clusters. (F) Expression of CD161, PD-1, TIGIT, and BTLA on total and relapse-associated CD4 T-cell clusters. (G) Expression of PD-1 and TIGIT on total and on the relapse-associated CD8 T-cell cluster. (H-I) Enrichment in the indicated IR positivity or expression density (median metal intensity, MMI) in relapse-associated clusters relative to total CD4 (H) and CD8 (I) T cells. (J) Frequencies of combined TIGIT clusters (C25+C26) or CD161 clusters (C22+C26) in relapsing (R) and nonrelapsing (NR) patients. Relapse-free survival (RFS) assessed using the Kaplan-Meier approach in patients with high (>median) vs low (<median) frequencies of indicated cell clusters are shown (right). P values by Mann-Whitney and log-rank tests, to compare cluster frequency and RFS, respectively. ****P < .0001.
CD161 and/or TIGIT are peculiar features of T-cell clusters associated with subsequent relapse
We then further deciphered what distinguished relapse-associated T-cell clusters from bulk CD4 or CD8 T cells. All of the identified clusters expressed PD-1 and at least 1 of the following receptors: TIGIT (C25, C26, C79), BTLA (C25, C26), and/or CD161 (C26, C22) (Figure 4F-G). To highlight specific features, we measured the enrichment in frequency or density of IR expression on the clusters vs bulk CD4 (Figure 4H) and CD8 (Figure 4I) T cells. Notably, although PD-1 was expressed on all cells of the 4 identified cell subsets, it was only poorly enriched because of substantial PD-1 expression on other T cells not associated with tumor relapse. In contrast, cell clusters had stronger enrichment in expression of either TIGIT (C25, C26) and/or CD161 (C26, C22), when measured as frequency of positive cells or as expression density (Figure 4H-I). Thus, TIGIT and/or CD161 expression appeared the most distinguishable features of T-cell clusters (non-MAIT) enriched in relapsing patients. Accordingly, higher levels of TIGIT+ (C25+C26) and of CD161+ (C26+C22) CD4 T-cell clusters at M3 after transplant were associated with shorter AML relapse-free survival (Figure 4J).
CD4 T cells expressing the noncanonical CD161 IR differ from cells with classic exhaustion phenotype and are strongly associated with poor relapse-free survival
Interestingly, CD161 was recently described as a novel IR restraining antitumor T cells in the context of malignant glioma where tumor cells expressed the CD161 ligand CLEC2D.35 Notably, in an ad hoc transcriptomic analysis using the TCGA Pan-Cancer Atlas Studies on cBioPortal,36 we found that AML ranked third of 35 cancer types with regard to CLEC2D expression levels (supplemental Figure 9).
Based on the results from the unsupervised clustering analysis described above, we next sought to further characterize total CD161+ but also TIGIT+ cells by classic gating in non-Treg CD4 T cells from healthy donors and patients of the cross-sectional cohort (Figure 5A). PD-1+ CD4 T cells were also studied as comparison. Although CD161 is highly expressed on MAIT cells, the analysis of CD161+ CD4 T cells did not include MAITs which are almost all CD4− (CD8+ or double negative).
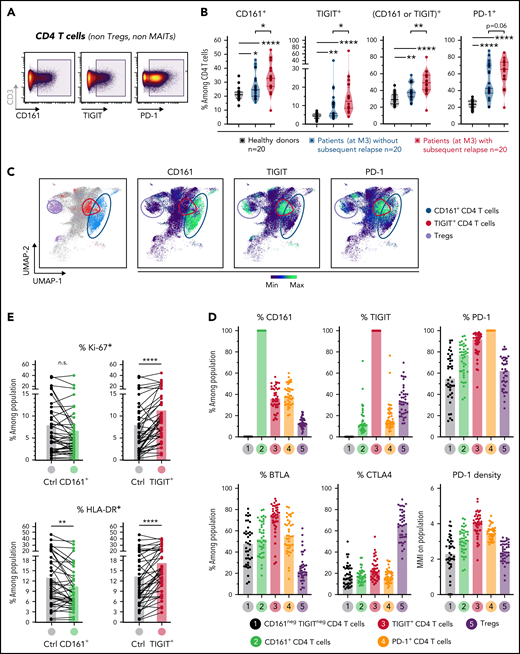
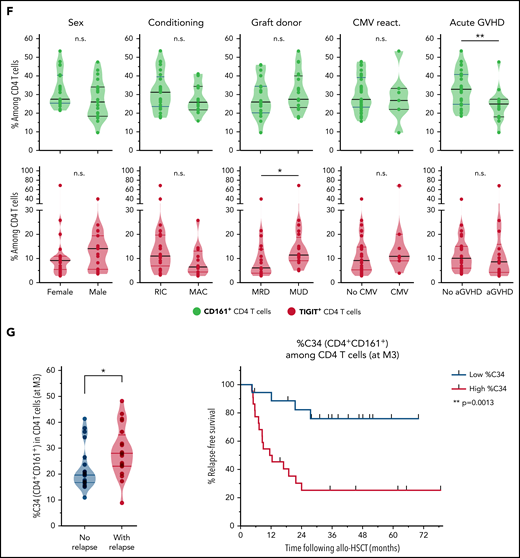
Characterization and clinical relevance of CD161+ and TIGIT+ CD4 T cells. (A) Representative gating of CD161+, TIGIT+, and PD-1+ CD4 T cells (after exclusion of Treg and MAIT cells). (B) Percentage of cells (non-Treg/non-MAIT) expressing CD161, TIGIT, at least CD161 or TIGIT (Boolean gating), or PD-1, among total CD4 T cells from healthy donors (n = 20), or from patients (at month 3 after allo-HSCT) with persistent remission (n = 20) or with subsequent tumor relapse (n = 20). (C) Uniform Manifold Approximation and Projection (UMAP) plots showing total CD4 T-cell clustering. Outlined cell subsets (from manual gating) are colored, whereas the ungated CD4 T cells are shown in gray (left). Expression levels of CD161, TIGIT and PD-1 are depicted (right), showing a poor overlap between CD161+ and TIGIT+ cells, but a strong overlap between TIGIT+ and PD-1high cells. (D) Frequencies of cells expressing various IRs among the indicated CD4 T-cell subsets. PD-1 cell surface density is also shown (expressed as median metal intensity, MMI) (bottom right). (E) Frequencies of HLA-DR+ and intranuclear Ki-67+ cells among CD161- and TIGIT-expressing CD4 T cells, compared with control (ctrl) cells that do not express CD161 and TIGIT. (F) Frequencies of CD161+ and TIGIT+ CD4 T cells were compared with regard to sex and various clinical parameters (ie, conditioning regimen, donor type, CMV reactivation, and acute GVHD). RIC, reduced intensity conditioning; MAC, myeloablative conditioning; MRD, matched related donor; MUD, matched unrelated donor. (G) FlowSOM clustering of PBMCs from the 40 patients at M3 after HSCT was performed based on lineage-defining markers (supplemental Figure 4). Frequencies of cluster 34 (left; corresponding to CD161+ CD4 T cells) among CD4 T cells, in nonrelapsing and relapsing patients. Relapse-free survival (RFS; right) assessed using the Kaplan-Meier approach in patients with high (>22%) vs low (<22%) frequencies of C34 in CD4 T cells. Censored patients are represented by symbols. P values by Mann-Whitney (B,F,G), Wilcoxon (E), or log-rank (G) tests: *P < .05; **P < .01; ****P < .0001.
Characterization and clinical relevance of CD161+ and TIGIT+ CD4 T cells. (A) Representative gating of CD161+, TIGIT+, and PD-1+ CD4 T cells (after exclusion of Treg and MAIT cells). (B) Percentage of cells (non-Treg/non-MAIT) expressing CD161, TIGIT, at least CD161 or TIGIT (Boolean gating), or PD-1, among total CD4 T cells from healthy donors (n = 20), or from patients (at month 3 after allo-HSCT) with persistent remission (n = 20) or with subsequent tumor relapse (n = 20). (C) Uniform Manifold Approximation and Projection (UMAP) plots showing total CD4 T-cell clustering. Outlined cell subsets (from manual gating) are colored, whereas the ungated CD4 T cells are shown in gray (left). Expression levels of CD161, TIGIT and PD-1 are depicted (right), showing a poor overlap between CD161+ and TIGIT+ cells, but a strong overlap between TIGIT+ and PD-1high cells. (D) Frequencies of cells expressing various IRs among the indicated CD4 T-cell subsets. PD-1 cell surface density is also shown (expressed as median metal intensity, MMI) (bottom right). (E) Frequencies of HLA-DR+ and intranuclear Ki-67+ cells among CD161- and TIGIT-expressing CD4 T cells, compared with control (ctrl) cells that do not express CD161 and TIGIT. (F) Frequencies of CD161+ and TIGIT+ CD4 T cells were compared with regard to sex and various clinical parameters (ie, conditioning regimen, donor type, CMV reactivation, and acute GVHD). RIC, reduced intensity conditioning; MAC, myeloablative conditioning; MRD, matched related donor; MUD, matched unrelated donor. (G) FlowSOM clustering of PBMCs from the 40 patients at M3 after HSCT was performed based on lineage-defining markers (supplemental Figure 4). Frequencies of cluster 34 (left; corresponding to CD161+ CD4 T cells) among CD4 T cells, in nonrelapsing and relapsing patients. Relapse-free survival (RFS; right) assessed using the Kaplan-Meier approach in patients with high (>22%) vs low (<22%) frequencies of C34 in CD4 T cells. Censored patients are represented by symbols. P values by Mann-Whitney (B,F,G), Wilcoxon (E), or log-rank (G) tests: *P < .05; **P < .01; ****P < .0001.
First, we found in a Boolean analysis that frequencies of CD4 T cells in patients at M3 after HSCT expressing (1) CD161+, (2) TIGIT+, and (3) at least TIGIT and/or CD161, were significantly elevated in patients with subsequent relapse (Figure 5B), whereas only a trend was observed for PD-1+ CD4 T cells. Of note, comparison with healthy donors showed that CD161 and TIGIT expression was upregulated in patients (Figure 5B) and remained stably elevated throughout the 1-year follow-up in the first longitudinal cohort (supplemental Figure 10). However, CD161 upregulation (median 1.3-fold increase vs healthy donors) was modest in comparison with the more markedly elevated expression of TIGIT (2.2-fold increase) and PD-1 (2.5-fold increase). Interestingly, Uniform Manifold Approximation and Projection (UMAP) embedding of CD4 T cells showed that CD161+ T cells overlapped poorly with TIGIT+ cells that clustered within PD-1–expressing cells (Figure 5C). Accordingly, TIGIT+ cells expressed higher levels of the other IRs PD-1 and BTLA (Figure 5D) and even higher PD-1 density than total PD-1+ cells themselves. Furthermore, TIGIT+ but not CD161+ cells were significantly enriched in dividing cells (intranuclear Ki-67 positivity) and in cells expressing HLA-DR, a marker of chronic T-cell activation (Figure 5E). Altogether, these data suggest that CD161+ cells, in contrast to TIGIT+ cells, exhibit a profile that is rather distinct from the classic exhaustion phenotype.
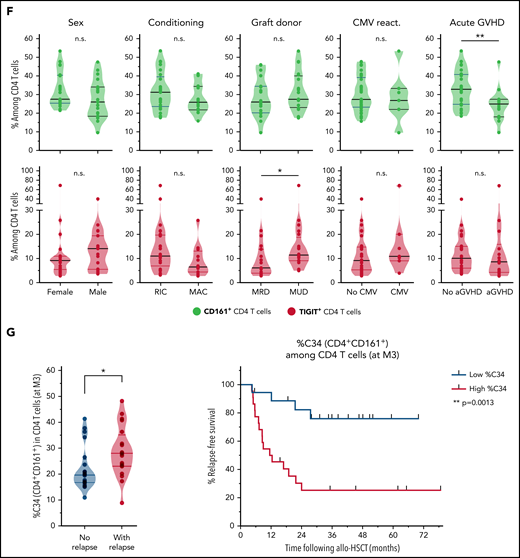
We then determined whether levels of CD161+ or TIGIT+ CD4 T cells may vary depending on various clinical parameters, including conditioning regimen, donor type, GVHD, and cytomegalovirus reactivation. Notably, no significant difference was observed for most parameters, except for increased frequencies of TIGIT+ cells in patients receiving transplants from unrelated donors and lower levels of CD161+ CD4 T cells in patients with acute GVHD (Figure 5F), more specifically, grade ≥3 aGVHD (supplemental Figure 11).
As TIGIT expression on not only CD4 but also CD8 T cells was associated with late relapse in the longitudinal cohort, we asked whether TIGIT+ T cells were associated with relapse-free survival in the cross-sectional cohort. Notably, high TIGIT expression on CD4 T cells and on naive, but not bulk, CD8 T cells was significantly associated with poor relapse-free survival (supplemental Figure 12).
Finally, in light of these results raising interest in CD161-expressing CD4 T cells, we further deciphered our previously mentioned FlowSOM clustering with phenotypical markers on bulk PBMCs (supplemental Figure 8A), because CD161 was included as a marker for MAIT cell identification. Interestingly, the cluster C34 corresponded to the total CD161+ (non-MAIT, non-γδ TCR+, and non-Treg) CD4 T-cell population (rather than a smaller FlowSOM subcluster as identified in Figure 4). Although C34 frequency among total PBMCs was not found to be related to relapse, we normalized its level to the CD4 compartment. Strikingly, high C34 cluster frequency among CD4 T cells was strongly associated with poor AML relapse-free survival (Figure 5G), thus confirming the clinical relevance of CD161 expression within CD4 T cells with regard to subsequent AML relapse.
Discussion
In this study, high-dimensional mass cytometry enabled us to comprehensively analyze the global immune reconstitution, focusing on the immunoregulatory landscape, after allo-HSCT in 77 patients with myeloid malignancies. We highlighted the overall relative dynamics and persistent alterations of a large range of immunoregulatory parameters. Importantly, a second specifically designed cohort study revealed, by both classic and unsupervised clustering analysis, that TIGIT and CD161 expression on CD4 T cells were distinct early parameters strongly associated with subsequent relapse.
The first cohort provided data for a global assessment of the longitudinal variations in distribution and phenotypic functional states of the main immune cell compartments during the first year after HSCT. Although there was substantial interindividual variability, the immune landscape was surprisingly stable between M3 and M6, whereas many significant changes occurred at M12. As previously demonstrated in studies focusing on individual cell subsets, the proportion of B cells and CD4 T cells was increasing at 1 year, at the expense of monocyte and NK-cell frequencies, reflecting the well-described reconstitution kinetics of these subsets.19,20 At 1 year, there was a persistent proportional lack of naive T cells, MAITs, ILCs (all subsets), and pDCs. Conversely, the NK cell compartment was disproportionate because of increased frequencies of cells with an immature CD56brightCD16− profile. Besides, nearly all T- and NK-cell subsets had higher expression of granzyme B and activation and exhaustion markers. This global approach thus highlighted that the overall immune landscape was still largely imbalanced, even 1 year after transplant. Of note, the data highlight a persistent general immune activation state, which may have implications for a long-term risk of cardiovascular comorbidities after HSCT,37 as has been well described for residual chronic immune activation in treated HIV infection.38
Regarding regulatory cell subsets, we found opposite patterns in Tregs and MDSCs dynamics. As did other CD4 T cells, Treg frequency increased and was biased toward the activated phenotype at 1 year after HSCT. In line with previous studies, this result may reflect the following: (1) thymic generation of Tregs, compared with other CD4 T cells, is markedly impaired after HSCT, so that Tregs in this context mostly expand by lymphopenia-driven homeostatic proliferation,39 and (2) naive Tregs have a strong potential to proliferate and convert to activated Tregs.32 These findings may thus account for the delayed increase in total Tregs and shrinkage of the naive Treg pool. In sharp contrast to Tregs, relative levels of monocytic, granulocytic, and early MDSC subsets were all abnormally elevated at M3 and decreased over time. Only sparse data are available about human MDSC subsets after allo-HSCT but, in line with our results, increased M-MDSCs levels were reported in this context.40 Of note, we found that only M-MDSC levels normalized at 1 year, whereas levels of G- and e-MDSC remained significantly elevated. The inflammatory context and severe immunosuppression after HSCT conditioning may be the driving forces for MDSCs expansion favored by inflammatory cytokines (eg, interleukin-6 or -1β) and emergency myelopoiesis.41,42 Of note, MDSCs may also originate directly from the graft, as these cells are enriched in peripheral blood grafts after granulocyte colony-stimulating factor stem cell mobilization.43 The strong and persistent lack of ILC reconstitution after allo-HSCT nearly rules out involvement of the ILC2-MDSC axis in this context.
We wanted to investigate whether immunoregulatory features after allo-HSCT may be associated with subsequent tumor relapse. In the first cohort including late relapse events, cell subsets associated with persistent remission interestingly mostly belonged to the innate-like T-cell compartment (MAIT, γδ T, and NKT cells) or conventional naive T cells. These cells mainly arise from de novo production in the thymus, which may stress the importance of the thymic function in the GVL effect. However, they all show different reconstitution dynamics, and innate-like T cells may also substantially depend on the commensal microbiota.44 These unconventional T-cell subsets with (semi)invariant TCRs may exert antitumor functions, although protumor roles have also been described (reviewed in Godfrey et al45). Their role in the context of HSCT is currently emerging, but studies have been limited by disease heterogeneity and the low number of patients.44 The first cohort in the present work also showed important limitations to study disease relapse (few and only late relapse events).
We thus specifically designed a homogeneous and balanced cohort matched for important confounding risk factors. Thereby, we identified via both classic and unsupervised clustering strategies, that early TIGIT and CD161 expression on CD4 T cells are associated with subsequent leukemia relapse. Higher PD-1 expression on CD8 T cells has been reported at the time of relapse after HSCT,7,8 which could be a cause but most probably a consequence of tumor development. However, exhausted PD-1+Eomes+T-bet− CD8 TSCM cells in the bone marrow early after transplant, as well as tumor-specific PD-1+ CD8 T cells, have been described in patients prone to relapse.8 Our work confirmed that PD-1–expressing T-cell clusters were associated with subsequent relapse. Nevertheless, we found that a more specific feature of T cells associated with relapse was the expression of a less commonly studied (TIGIT) and a newly identified (CD161) IR.35 Elevated TIGIT expression on CD8 T cells was reported in patients with AML and corelated with poor clinical outcome.46 Importantly, the TIGIT ligands CD155/PVR and CD112/PVRL2 are expressed on AML blasts,7,46,47 and percentage and/or expression density of these receptors are even increased on AML blasts at relapse after HSCT, compared with those at diagnosis. Together with our results, data argue for the involvement of TIGIT-mediated immune escape in this context.
Of note, DNAM-1 (CD226) share the same nectin-like ligands as TIGIT, but is conversely an activating receptor involved in T-cell– and NK-cell–mediated cytotoxicity. Interestingly, contrary to TIGIT, DNAM-1 expression is decreased on T and NK cells from patients with AML.46,48 One could hypothesize that concomitant loss of DNAM1 together with TIGIT upregulation is an exhaustion phenotype that may play a role in AML relapse, as shown in a murine model of myeloma escape after autologous HSCT.49
Although CD161 is a classic marker for MAIT cells, unsupervised analyses unexpectedly pinpointed the clinical relevance of CD161 expression on CD4 T cells that were neither MAIT nor Treg or γδ T cells. Of note, CD161-expressing CD4 T cells may encompass Th17 cells, but these are rare compared with total CD161+ CD4 T cells. Interestingly, CD161 was very recently described as a novel IR restraining antitumor CD8 and CD4 T cells in glioma.35 CD161 and TIGIT expression poorly overlapped and CD161+ CD4 T cells were more distinct from a classic exhaustion phenotype than TIGIT+ CD4 T cells. As high level of CD161+ CD4 T cells was strongly associated with poor relapse-free survival (and also with absence of severe acute GVHD), the functional role of this molecule will deserve further investigation in this context. Importantly, the CD161 ligand CLEC2D (LLT1) showed one of the highest expression levels in AML when compared to a large range of cancer types (including glioblastoma).
Interestingly, we found an association between relapse and IR expression mostly within the CD4 T-cell compartment. Notably, it had been demonstrated that (1) MHC class II (rather than class I) molecules are downregulated on AML blasts at relapse vs at diagnosis,7,9 (2) HLA-DP mismatches reduce the risk of leukemia relapse,50 and (3) CD4 T cells are necessary, and possibly sufficient, for GVL responses in murine models of donor lymphocyte infusion.51,52 Altogether, these data suggest a crucial role for effective CD4 T cells in the GVL effect, as cytokine-secreting helper cells but also, possibly, as direct cytotoxic lymphocytes.53
In the present study, neither Tregs nor any MDSCs subsets were associated with subsequent relapse. This finding is of interest for strategies to increase their number to prevent or treat GVHD while preserving the GVL effect. Indeed, besides ongoing efforts on Treg cell therapies, MDSCs also inhibited GVHD in preclinical models,54 and ILC2 infusion has also raised interest in inducing tolerance via MDSC induction.18,55
Limitations of this work include the lack of data regarding the bone marrow immunoregulatory landscape or regarding diversity of the TCR repertoire and T-cell metabolism that may also be of interest with regard to the GVL effect.56-58 Moreover, this study was limited to HLA-matched transplantation to obtain a homogeneous cohort, thus limiting confounding factors, but more recent approaches such as haploidentical HSCT may warrant further investigations. Notably, in the latter setting, impaired NK cell recovery after posttransplant cyclophosphamide was found to predict relapse.59 Finally, what factor(s) may affect TIGIT and CD161 expression levels, which were highly heterogenous between patients but rather stable over the months, remains an open question. It would be of interest to assess whether these are correlated with the levels found in the corresponding graft donors.
Overall, there is now a growing body of evidence arguing for IR-mediated AML immune escape after allo-HSCT. Yet PD-1 blockade is the most commonly used immunotherapy to date, one could speculate that TIGIT and CD161 may represent more relevant targets in this context. The use of immune checkpoint-blocking antibodies may raise concerns about exacerbating GVHD, so that innovative therapeutic strategies could be necessary to selectively target and reinvigorate tumor-specific but not healthy tissue-specific alloreactive T cells.
Acknowledgments
The authors thank Elise Diaz and Marc-Antoine Silvestrini (INSERM U976) for help in editing the manuscript; all the patients, their physicians, the nurses, and technicians from Hôpital Saint-Louis, who helped with the study; and CRYOSTEM for cell collection management.
This work was supported by grants from Fondation pour la Recherche Médicale (FRM) and Cancéropôle Île-de-France, Institut National du Cancer (INCa) (M.F.C.). V.G. was supported by a fellowship from Agence Régionale de la Santé (ARS Île-de-France).
Authorship
Contribution: M.F.C. conceived the study, analyzed the data, and wrote the manuscript; V.G., V.P., N.V., and K.V. conducted the experiments and analyzed data; A.C., J.B., M.L., and D.M., participated in collecting experimental data, developing the methodology, and editing; V.G., R.P.d.L., and G.S provided patient samples and data; V.G., S.C.-Z., and G.S. discussed the results and edited the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mathieu F. Chevalier, INSERM U976, Hôpital Saint-Louis, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: mathieu.chevalier@inserm.fr; and Gérard Socié, Hématologie/Transplantation, Hôpital Saint-Louis, 1 Ave Claude Vellefaux, 75010 Paris, France; e-mail: gerard.socie@aphp.fr.
Original data are available by e-mail request to one of the corresponding authors.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.