IN UTERO HEMATOPOIETIC stem cell transplantation (IUHSCTx) is a theoretical alternative to postnatal stem cell transplantation (SCT) for the treatment of congenital hematologic disorders that can be cured by SCT, and can be diagnosed early in gestation. Advances in prenatal diagnosis and molecular analysis now allow diagnosis of the majority of congenital hematologic disorders by10 to 12 weeks’ gestation. The evolution of high-resolution ultrasound and precise interventional techniques have solved the technical obstacles to performing early gestational cellular transplants. Considering these advances, it stands to reason that there is increasing clinical interest in performing IUHSCTx and that there will inevitably be an increasing number of attempts to treat fetuses with hematologic disorders in utero.
The rationale for consideration of IUHSCTx is based on normal developmental ontogeny. The early gestational fetus is immunologically immature and uniquely tolerant to foreign antigen, allowing acceptance of allogeneic or xenogeneic cells without the need for immunosuppression. Under specific circumstances, the fetal environment appears permissive to engraftment of HSC without the requirement for myeloablation. The maternal womb is the ideal sterile isolette, allowing the potential for immunologic reconstitution before birth. Finally, successful prenatal transplantation could preempt clinical manifestations of the disease, avoiding the need for postnatal treatment and the high cost in human suffering, and expense to society, currently associated with SCT.1 2 This is the clinical promise of IUHSCTx. To date, this promise remains unfulfilled. With increasing experimental and clinical experience, the naive concept that a simple transplant in utero might cure a large number of diseases has given way to a realistic appreciation of the obstacles to successful engraftment. Reality has forced reconsideration of the original assumptions about fetal transplant biology, and resulted in formulation of new questions. It has also resulted in the consideration of new strategic approaches for the therapeutic application of IUHSCTx, in a variety of clinical circumstances.
“NATURAL” HEMATOPOIETIC CHIMERISM
The best supporting evidence that IUHSCTx might work remains an “experiment of nature” first described by Owen in 1945.3 He observed that dizygotic cattle twins that share cross-placental circulation were born chimeric for their siblings’ blood elements. This state of “mixed chimerism” persists for life and is associated with specific transplantation tolerance.4,5 Natural chimerism has been observed in other species as well, most notably, humans6,7 and the cotton-top tamarin (primate).8 9 Interestingly, it has been observed that donor hematopoiesis in some chimeric animals can actually predominate, with the persistence of very high levels of donor-derived cells. This experiment of nature represents “proof in principle” that, under specific circumstances, allogeneic donor cells can competitively populate a hematopoietically normal recipient, with substantial and stable levels of donor cell expression.
EXPERIMENTAL HEMATOPOIETIC CHIMERISM AFTER IUHSCTx IN NORMAL ANIMAL MODELS
Efforts to reproduce “natural” chimerism in the laboratory by the prenatal transplantation of allogeneic or xenogeneic HSC have had variable degrees of success. The most successful animal model remains the sheep. Early gestational transplantation of allogeneic, fetal liver–derived HSC into normal sheep fetuses results in a high rate of sustained multilineage hematopoietic chimerism10 that persists for many years and is typically in the range of 10% to 15% bone marrow (BM) and peripheral blood donor cell expression.11 The fetal sheep model is also permissive for widely disparate xenogeneic engraftment. Multilineage hematopoietic chimerism has been well documented after human fetal liver–derived HSC transplantation12 and after transplantation of a variety of human cord blood and adult BM-derived populations.13-18 In addition, we have shown that chimerism in the human sheep model is caused by the engraftment of pluripotent HSC by documentation of long-term engraftment by donor cells on retransplantation into second-generation fetal lamb recipients.19 In contrast to the sheep, however, other normal animal models have shown much greater resistance to engraftment after in utero transplantation. Although chimerism has been achieved in the normal primate,20goat,21 rat,22 and mouse,23-26 the levels of engraftment are much lower and well below what might be expected to be therapeutic for most hematologic diseases.
EXPERIMENTAL HEMATOPOIETIC CHIMERISM AFTER IUHSCTx IN IMPAIRED ANIMAL MODELS
In contrast to normal animal models, it is clear that under circumstances where there is a competitive advantage for normal cells, high levels of donor cell engraftment can be expected. This was first shown by Fleischman and Mintz27 in studies in W mutant anemic mouse strains that have a stem cell deficiency based on the absence of c-kit. In utero transplantation of normal allogeneic fetal liver cells by transplacental injection at 11 days gestation resulted in rescue of severely anemic mice and complete reconstitution by donor hematopoiesis. The degree of erythroid replacement correlated with the degree of underlying anemia, with complete early replacement by donor erythroid cells in the lethally anemic W/W homozygotes, and partial but progressively increasing replacement by donor erythroid cells in the sublethally anemic Wv/Wvhomozygotes. Donor white blood cell (WBC) engraftment was also seen in the Wv/Wv recipients, but was not as extensive as erythroid engraftment, mirroring the underlying severity of the lineage defect. In a less severe model of anemia based on a different mutation of c-kit (W41/W41),28 Blazar et al29documented high levels of multilineage chimerism of congenic donor cells with confirmation of HSC engraftment by repopulation of irradiated secondary recipients. Similarly, in the mouse severe combined immunodeficiency disease (SCID) model in which there is early arrest of T- and B-cell development, Blazar et al30 have demonstrated lymphoid reconstitution after IUHSCTx. In successfully reconstituted animals, T and B lymphocytes were entirely of donor origin. Although donor myeloid and erythroid elements could not be consistently detected, the engraftment of donor HSC in the marrow was clearly documented by retransplantation experiments. Thus, in the presence of a lineage deficiency, IUHSCTx can selectively reconstitute the defective lineage, but it appears that competitive pressure from the normal host lineages prevents high-level multilineage donor cell expression. More recently, engraftment was compared in the SCID mouse model following IUHSCTx versus nonconditioned postnatal SCT.31 There were a number of advantages favoring IUHSCTx found in this study, including a lower risk of graft-versus-host disease (GVHD) and more rapid and earlier lymphoid reconstitution of the thymic and splenic compartments. Recent studies in the nonobese diabetic (NOD)/SCID mouse confirm and expand upon these observations.32 In this model, the defect in T- and B-cell development is the same as the SCID mouse but in addition there are known defects in natural killer (NK) cells and antigen presentation.33 IUHSCTx in NOD/SCID recipients results in multilineage engraftment with increasing donor cell expression over time.
CLINICAL EXPERIENCE WITH IUHSCTx
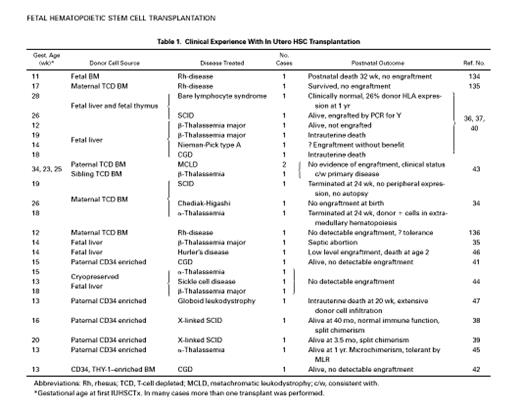
There have now been 26 human cases of IUHSCTx that have been reported in the literature or are personally documented by the authors. There have also been a significant number of attempts that are either pending or have not been reported that cannot be commented upon in this review. Transplants have been performed by numerous investigators, for many different diseases, using a variety of transplant protocols. The authors have recently reviewed the reported clinical experience with IUHSCTx2 and this is updated in Table 1. It is important to note that of the 20 non-SCID cases, 12 have been performed beyond 14 weeks’ gestation, when the human fetus would be expected to be immunocompetent. In addition there have been at least 3 early deaths, 1 from sepsis (fetal liver source) and 2 procedural that should, in the modern era, be avoidable. Two cases (1 SCID and 1 α-thalassemia) underwent umbilical blood sampling at 24 weeks, and termination was performed when no peripheral donor cell expression was found.34 35 We now know that peripheral expression of engrafted donor cells does not occur until near term and it is interesting that at autopsy, the α-thalassemia fetus had donor cells detected in multiple areas of extramedullary hematopoiesis.
The only clear successes, or claims of success, have been in immunodeficiency disorders in which there is a selective advantage for donor cells.36-39 It is clear from these reports that X-SCID can be effectively reconstituted by paternal CD34-enriched BM. Our patient is now 4 years old and maintains excellent cellular reconstitution and has shown the ability to specifically respond to vaccinations, including the novel T-cell antigen φX174, which requires intact T-cell help for B-cell Ig class switching (unpublished data, June 1999). In other immunodeficiency disorders, such as chronic granulomatous disease (CGD) (3 cases)40-42or Chediak-Higashi Syndrome,34 no engraftment was observed at birth. Two recent CGD cases were of particular interest because they failed, despite early gestational transplantation and the use of either the same protocol utilized for successful treatment of X-SCID,41 or transplantation of more highly HSC-enriched donor cells.42
The treatment of hemoglobinopathies by IUHSCTx has thus far been unsuccessful. There have been 5 cases of β-thalassemia attempted, of which 2 were intrauterine deaths.35,40,43,44 The 3 others were transplanted at 12,18, and 25 weeks’ gestation. The transplant at 12 weeks used fetal liver as a donor source and was initially reported as being engrafted with polymerase chain reaction (PCR), evidence of the presence of Y chromosome and HbA levels of 0.9% at birth, which subsequently increased to 30% at 1 year of life. This patient has since been reported to have lost engraftment and remains transfusion dependent.37,40 There have been 3 reported attempts to treat α-thalassemia by IUHSCTx.34,44,45 One used cryopreserved fetal liver as a donor source at 15 weeks’ gestation and failed. One had evidence of donor cell engraftment in areas of extramedullary hematopoiesis, but was terminated because of lack of peripheral donor cell expression at 24 weeks. The third received the same protocol of CD34-enriched paternal BM successfully used for X-SCID, with the first transplant administered at 13 weeks’ gestation. The fetus required blood transfusions for fetal anemia and had only microchimerism of donor cells detected at birth. This patient was, by report, tolerant to donor antigen by mixed lymphocyte reaction. Only one attempt has been reported for sickle cell disease.44 Cryopreserved fetal liver was used at 13 weeks’ gestation and no detectable engraftment was noted after birth.
Finally, 5 attempts have been reported to treat fetuses with metabolic storage diseases by IUHSCTx.40,43,46,47 Two of these were performed too late in gestation (34 and 23 weeks) to expect engraftment, and none was observed. One was performed for Nieman-Pick Type A at 14 weeks and the engraftment status is unclear, but there was no clinical benefit. A patient with Hurler’s disease was transplanted using fetal liver and had evidence of low-level engrafment with evidence of increasing enzyme production, but had severe clinical manifestations of the disease, with death at age 2. In the third case, an attempt was made to transplant a fetus with globoid leukodystrophy with a higher dose of CD34-enriched BM cells. Adequate T-cell depletion was not performed, and in excess of 107 CD3+cells/kg fetal weight were delivered to a 13-week gestation fetus. The fetus died at 20 weeks’ gestation, with autopsy findings in an autolyzed fetus of “overwhelming myelopoiesis” as documented by myeloperoxidase staining.47 A more likely cause of death was GVHD, which has not been convincingly excluded by the authors.
In total, this limited experience supports the presence of significant barriers to engraftment in human fetuses after IUHSCTx in diseases where there is little or no selective advantage for normal cells. The nature of this barrier is poorly understood.
THE ENGRAFTMENT BARRIER
The primary determinant for expanding the clinical application of IUHSCTx will be the ability to improve engraftment in the hematopoietically competitive recipient. To improve engraftment in competitive prenatal environments, the unique transplantation biology of the prenatal recipient must be better defined and the barriers to engraftment identified. IUHSCTx differs in 3 major respects from postnatal SCT. First, there is abundant evidence that the myeloablative regimens and irradiation used to permit engraftment after postnatal SCT alters the biology of the recipient hematopoietic microenvironment.48-53 Second, after IUHSCTx, there is competition from a preexisting, vigorous, host hematopoietic compartment that is not present after myeloablation. Third, there is the underlying framework of normal hematopoietic and immunologic ontogeny. Therefore, the paradigm of postnatal SCT does not necessarily apply to IUHSCTx. Insight into the barriers to prenatal engraftment can only be obtained by consideration of biologically relevant competitive model systems. Figure 1schematically depicts the models and contrasts the competitive circumstances for donor cells. The following discussion summarizes what, in the authors’ view, is the current experimental evidence derived from these models that is directly relevant to IUHSCTx. This evidence will be presented in the context of 3 assumptions that have been used in the past as a presumptive basis for IUHSCTx.

Assumption no. 1: There is “space” in the expanding fetal hematopoietic compartment that is available for homing and engraftment of donor cells.
During fetal development, there is sequential migration of hematopoiesis from the yolk sac and/or para-aortic splanchnopleure to the fetal liver and, subsequently, to the BM.54,55 There is an associated exponential expansion of the hematopoietic compartment with presumably continuous formation of new microenvironmental sites, or “niches,” for homing and engraftment of circulating HSC. We and others have suggested that the number of “niches” available for engraftment in the prenatal microenvironment probably exceeds the availability of niches in the postnatal environment, offering one explanation for the ability to engraft fetal recipients without myeloablation. That precept has been challenged by a number of recent prenatal and postnatal observations. The dogma that the creation of “space” is required for the engraftment of donor cells after birth has been increasingly challenged, primarily on the basis of data derived from the syngeneic nonmyeloablated mouse model, originally described by Brecher et al56 and recently revitalized by Stewart et al.57 In this model system, analogous to IUHSCTx, there is no irradiation effect and the host hematopoietic compartment is intact. However, it differs from allogeneic IUHSCTx in that engraftment occurs in the postnatal BM environment and donor and recipient cells are genetically equal and syngeneic with stromal elements.
In this model, stepwise increases in donor cell engraftment can be achieved with repetitive large doses (1 to 2 × 109cells/kg) of syngeneic donor BM cells.57,58 This suggests that there is a steady state of open receptive sites in normal bone marrow. More recent interesting observations in this model include the observation that no saturation point could be demonstrated with increasing doses of donor cells (up to a dose of 1 to 2 × 109 cells/kg administered on 5 consecutive days), and that a single overwhelming dose of cells (0.5 to 1 × 1010cells/kg) resulted in equivalent engraftment at 7 to 14 weeks after transplantation.59 The observations of no saturation point and equal engraftment with overwhelming doses of cells support actual displacement of host cells by an excess of circulating donor cells. In this model, donor cell engraftment appears quantitative at the stem cell level so that peripheral donor cell expression is determined by the ratio of donor to host HSC. This is in keeping with observations on stem cell kinetics in nonirradiated systems. Whereas in the irradiated mouse or large animal it is well documented that engraftment of a single or few HSC can provide oligoclonal reconstitution, providing an “amplified” readout of engraftment,60,61 studies of stem cell kinetics in normal mice62-64 and allophenic mice65-67 suggest that nearly all HSC regularly cycle and that the sum of hematopoiesis is provided by many simultaneously cycling HSC. Thus, engraftment in this system of a single or few HSC would be relatively difficult to detect, corresponding to the observations in the in utero models (Fig 2).

Comparison of engraftment characteristics in various hematopoietic systems that differ in their donor and host HSC competitive capacity, assuming a model of relatively frequent stem cell cycling in nonirradiated systems. (A) Irradiated postnatal environment. Damaged microenvironment results in engraftment of a few donor HSC that reconstitute the recipient by oligoclonal expansion, allowed by the absence of host cell competition. There is an amplified “readout” of a few engrafted donor HSC. (B) Allogeneic IUHSCTx. Although a few donor HSC may engraft, they are at best equal, or more likely, at a competitive disadvantage to host HSC (mismatched stroma). Therefore, the engraftment is obscured by host hematopoiesis leading to a readout of minimal or microchimerism. (C) The syngeneic nonmyeloablated model. Engraftment through serial transplants or massive doses of donor cells results in multiple-donor HSC engrafted that can equally compete (genetically identical HSC/matched stroma) with host HSC. Engraftment readout is quantitatively reflective of No. Donor HSC/No. Host HSC. (D) Allogeneic IUHSCTx into a host with impaired HSC. Only a few donor HSC engraft, but oligoclonal expansion can occur despite stromal mismatch, due to reduced host HSC competition, leading to an amplified readout of donor HSC engraftment.
Comparison of engraftment characteristics in various hematopoietic systems that differ in their donor and host HSC competitive capacity, assuming a model of relatively frequent stem cell cycling in nonirradiated systems. (A) Irradiated postnatal environment. Damaged microenvironment results in engraftment of a few donor HSC that reconstitute the recipient by oligoclonal expansion, allowed by the absence of host cell competition. There is an amplified “readout” of a few engrafted donor HSC. (B) Allogeneic IUHSCTx. Although a few donor HSC may engraft, they are at best equal, or more likely, at a competitive disadvantage to host HSC (mismatched stroma). Therefore, the engraftment is obscured by host hematopoiesis leading to a readout of minimal or microchimerism. (C) The syngeneic nonmyeloablated model. Engraftment through serial transplants or massive doses of donor cells results in multiple-donor HSC engrafted that can equally compete (genetically identical HSC/matched stroma) with host HSC. Engraftment readout is quantitatively reflective of No. Donor HSC/No. Host HSC. (D) Allogeneic IUHSCTx into a host with impaired HSC. Only a few donor HSC engraft, but oligoclonal expansion can occur despite stromal mismatch, due to reduced host HSC competition, leading to an amplified readout of donor HSC engraftment.
In the fetus, we have assumed that “space” would be relatively available due to the rapid expansion of the fetal hematopoietic compartment that, by necessity, must include the rapid formation of new niches. In reality, however, the availability of niches for donor cell engraftment would depend on the dynamic balance of stromal and hematopoietic cell expansion. What is the balance of stromal receptive sites and circulating hematopoietic progenitors in the fetus? There is limited direct data available. In a developmental study of stromal (CFU-F) and hematopoietic elements in the fetal liver and BM, Wolf et al68 observed that stroma formation preceded hematopoiesis in the fetal liver (at 13 days) and BM, but that hematopoietic activity increased very rapidly after the establishment of stroma. It has been well documented that the number of HSC and progenitors circulating in fetal peripheral blood is much higher than the number in cord blood, or after birth, supporting relative HSC excess.69 Thus, there is little reason to expect that once a niche forms in the fetal environment it will remain available for donor cell engraftment.
There are also a number of direct observations that support limited host receptivity as a barrier to engraftment in utero. In the fetal lamb model using allogeneic or xenogeneic fetal liver– or BM-derived donor cells, log-fold increases in donor cell dose (106 to 1010 cells/kg) increase engraftment to some extent, but a plateau is reached where further increases in donor cell dose have no effect.70 This suggests that available receptive sites can be saturated with donor cells, limiting further increases in donor cell engraftment. In contrast to the syngeneic, nonmyeloablated mouse model, the ability of overwhelming doses of donor cells to “displace” host cells in the fetus has not yet been shown. Also in the fetal lamb model, transplantation of divided doses of donor cells at intervals increases engraftment significantly above that achieved by transplantation of the same number of cells in a single dose.46 This observation is identical to the results in the syngeneic nonmyeloablative mouse model and suggests that new receptive sites form, or become open, in the time interval between transplants to allow engraftment of additional donor HSC. Thus, the available evidence suggests that there is not an abundance of space available in the fetal microenvironment, relative to the postnatal BM microenvironment, and that a limited number of receptive sites is at least one component of the barrier to engraftment in normal fetal recipients.
Assumption no. 2: Donor HSC can effectively compete with host HSC to achieve significant donor cell expression after IUHSCTx.
By this assumption, successful reconstitution is dependent on the ability of an initial inoculum of donor HSC to survive and expand into the host hematopoietic space. If donor cells have a competitive advantage, then even the engraftment of a relatively limited number of donor HSC could ultimately reconstitute the recipient. In normal animal models, we have observed after IUHSCTx little evidence that donor cells can expand their presence in the host milieu, except in the human/sheep model when a competitive advantage is conferred by infusion of donor species-specific human cytokines.12,71 In contrast, there is abundant evidence that in circumstances of donor cell competitive advantage, donor cells rapidly expand into a deficient compartment. The high levels of donor hematopoiesis achieved in c-kit–deficient mouse strains in which there is a proliferative defect in host HSC support this hypothesis. Mintz et al72 have documented full reconstitution in this model after IUHSCTx by 1 or 2 normal HSC. In a separate study, Fleischman73 showed that when donor cells have equivalent c-kit function to host cells, W mutant mice do not accept grafts more readily than wild-type animals, supporting a competitive advantage, rather than space, as the primary determinant of donor cell expression.
Experimental evidence also supports the ability of limited numbers of donor HSC to fully reconstitute specific defects in host lineage development. In the mouse SCID model30 as well as in a human X-SCID patient,38 the number of engrafted HSC in the BM remains relatively low despite full reconstitution of the defective lineage. In this system, there is no selective advantage at the HSC level. Thus, the number of donor HSC do not appear to expand over time, consistent with a mechanism of reconstitution by expansion of lineage-committed cells that are replenished from a limited pool of stem cells.
In another recent study, the syngeneic nonmyeloablation model was modified by exposure of the host to minimally myeloablative radiation (100 cGy). Syngeneic donor cells showed high levels of donor cell engraftment despite transplantation of relatively low numbers of cells.74 Transplantation of donor cells receiving the same dose of radiation reduced donor cell engraftment to 14% of that seen with nonirradiated cells, strongly supporting the concept that it is primarily the ability of host cells relative to recipient cells to compete, rather than space, that determines ultimate engraftment.
Another model system that is highly analogous to the biology of IUHSCTx is the allophenic mouse.75 In this model, allogeneic cells coexist, in an immunologically tolerant system, from the embryonic stage forward. As in IUHSCTx, allogeneic cells may have genetic differences in their competitive capacity; however, there is complete developmental mixing of the cells from day 2 of gestation and, more importantly, all cells, including stroma, thymus, and other tissues, are chimeric. In allophenic mice in which one strain has an HSC pool with relatively rapid cycling kinetics (DBA/2 vC57BL/6),76 the early kinetics of engraftment favor DBA/2 stem cells until they become senescent and C57BL/6-derived hematopoiesis becomes predominant.65 The allophenic studies show that in a chimeric microenvironment in which allogeneic cells compete, the level of expression is a function of the genetically defined competitive capacity of the HSC.
However, the ability of donor cells to compete is not purely HSC derived. A number of recent findings suggest that in nonirradiated systems, HSC stromal interaction is at least partially MHC restricted. Transplantation of a donor microenvironment (bone containing intact stroma) allows engraftment of donor HSC in usually resistant strain combinations.77,78 In a compelling study, Hashimoto et al79 transplanted donor autologous microenvironment (bone) and major or minor major histocompatibility complex (MHC) mismatched microenvironment into irradiated allogeneic recipients. Observations included the following: (1) increased cellularity and number of progenitors in MHC-matched BM versus recipient or third-party BM; (2) the ability to engraft HSC when combined with the autologous, but not mismatched, microenvironment into previously resistant strain combinations; (3) the dependence of this advantage on MHC with no restriction demonstrated for minor antigen disparity; and (4) MHC restriction was abrogated by pretransplant irradiation of the donor microenvironment. In addition, in other studies autologous but not MHC disparate “facilitator cells” are effective in enhancing engraftment.80 81 In combination, these studies support the concept that in nonirradiated recipients, donor cells must have their own supporting stromal elements to optimally compete with recipient cells.
In summary, there is clearly overlap between the hypotheses that available space and competitive capacity limit donor cell engraftment. Data suggest that if donor and host cells are truly competitively equal, donor cell expression will quantitatively reflect the ratio of donor to host HSC. Thus, a large number of HSC would need to be engrafted to provide clinically significant levels of donor cell expression, and the number of receptive sites may be a critical limitation. However, under circumstances of even minimal imbalance favoring host hematopoiesis, the primary barrier to donor cell engraftment and expression will be host cell competition. Thus, it would appear that after IUHSCTx, donor hematopoiesis is limited by both an inability to engraft an adequate number of donor cells, because of a lack of receptive sites, and the subsequent inability of this limited number of engrafted cells to expand into the host hematopoietic compartment, because of what is probably a competitive disadvantage.
Hypothesis no. 3: The early gestational fetus is immunologically tolerant of foreign antigen.
Since Billingham et al’s82 classic observations of “acquired” immunologic tolerance, the phenomenon of fetal tolerance has been relatively accepted. Evidence is now overwhelming that the fetal thymic microenvironment plays a primary role in determination of self recognition and repertoire of response to foreign antigen. Pre-T cells undergo positive and negative selection during a series of maturational steps in the fetal thymus that are controlled by thymic stromal cells.83,84 The end result is deletion of T-cell clones with high affinity for self antigen in association with self-MHC, and preservation of a T-cell repertoire against foreign antigen. Therefore, theoretically at least, introduction of foreign antigen before thymic processing should result in presentation of donor antigen in the thymus with clonal deletion of alloreactive T cells. However, it is important to note that the mechanism of central thymic tolerance has been defined primarily in T-cell receptor (TCR) transgenic mice. In these mice, thymic maturation of lymphocytes occurs in an environment of unregulated high levels of TCR with high affinity for a specific self antigen, which is expressed from the earliest to the latest stages of thymic development.85-87This is distinct from the clinical situation after IUHSCTx in which there are a large number of circulating antigens interacting with recipient TCRs of varying affinity for donor antigen. Differences in thymic maturation of lymphocytes in normal mice from the defined mechanisms in TCR transgenic mice have been recognized.88,89 In addition, there are other mechanisms of rejection including NK- or B-cell–mediated response that are relatively poorly understood. In fact, experimental efforts to induce tolerance by prenatal presentation of antigen have had inconsistent results. In the initial report of Billingham et al,82 only 3 of 5 survivors were tolerant of donor skin grafts (in an MHC class I disparate but class II matched strain combination), and in many other investigations, particularly in xenogeneic combinations, results have been inconsistent.90-92 These classical studies are difficult to interpret because no analysis of donor cell chimerism could be performed. In more recent studies, we failed to demonstrate specific tolerance induction for allogeneic renal grafts in recipient lambs made chimeric by in utero transplantation of T-cell–depleted adult marrow, despite the measured presence of 2% to 5% donor hematopoietic engraftment.93 Carrier et al24documented specific tolerance to skin grafts in only 3 of 22 mice with microchimerism after fully allogeneic IUHSCTx. In this same microchimeric model we have shown that tolerant animals exhibit a combination of partial clonal deletion and clonal anergy of residual donor reactive cells, whereas in nontolerant animals no evidence of deletional tolerance is present.94 In general, these studies support the existence of the phenomenon of fetal tolerance but suggest that it may be conditional and dependent on timing and appropriate presentation of antigen in the fetus.
OVERCOMING THE ENGRAFTMENT BARRIER
Consideration of IUHSCTx in the context of the above discussion suggests a number of strategies by which higher levels of engraftment in competitive systems might be achieved. These strategies fall into the category of either increasing the number of donor HSC engrafted or increasing the competitive capacity of donor-derived hematopoiesis relative to host hematopoiesis. Strategies designed to increase the number of donor HSC engrafted must assume a model of at least equal competitive capacity of the donor cells after engraftment. In this model, the level of engraftment would be equal to the fractional representation of donor HSC in the host environment. By this model, it is clear that far higher numbers of HSC would need to be engrafted than are currently engrafted after IUHSCTx. Direct approaches of increasing the number of donor HSC by increasing cell number or HSC enrichment have been tested to a limited extent in animal models without dramatic increases in engraftment. However, it is fair to say that the upper limits of this strategy have not been explored in fetal models, and if host cells can be “displaced” by massive doses of donor cells,59 such a strategy might be successful. The clinical limitation to this strategy when using adult BM as a donor cell source is the T-cell dose administered with higher numbers of CD34+ cells.47 Another direct approach is to perform multiple transplants in hopes of maintaining circulating levels of donor cells to engraft as niches form or become available. This strategy has been highly successful in the sheep with significant increases in engraftment even when the same absolute numbers of cells are given.46 Multiple prenatal transplants have also been given clinically with success in X-SCID,38,39 where there is a selective advantage for normal cells, but no appreciable engraftment was achieved using similar protocols in CGD or α-thalassemia.2 45 Other approaches, such as selective strategies to improve homing and engraftment of donor HSC or the use of specific populations of donor cells with optimal engraftment characteristics, may be useful but await experimental support. Finally, methods to minimally ablate the fetus to increase the number of receptive sites have, in general, not been investigated and would require absolute assurances of safety and absence of long-term morbidity before clinical application.
As delineated above, there is no reason to believe that engraftment of more HSC would necessarily lead to higher levels of engraftment. The weight of evidence suggests a model for engraftment following IUHSCTx in which there is not only limited receptivity to engraftment, but also a competitive disadvantage for donor cells. In this circumstance, engraftment of even a large number of donor cells would be overwhelmed by host hematopoiesis. Strategies to improve engraftment assuming this model generally depend on improving the relative competitive capacity of the donor cell population. Once again there is overlap between the models. The use of donor-specific stromal cotransplantation has been experimentally promising in the sheep model and significantly increases short- and long-term donor cell expression.96 Whether the improvement in engraftment is due to BM “conditioning” to provide more receptive sites, or to improved competition by donor cells caused by their interaction with matched stromal elements is unknown. However, even if competitive capacity were equalized, clinically significant donor cell expression would still require the engraftment of higher numbers of donor HSC than have generally been achieved after experimental or clinical IUHSCTx. On the other hand, strategies that would provide an actual competitive advantage for donor cells would theoretically be effective with engraftment of even a few donor cells, because they would be able to expand into the host compartment. It should be emphasized that the strategy of prenatal tolerance induction, by establishment of minimal chimerism, followed by postnatal boosting of engraftment using minimally ablative regimens, would theoretically not only engraft more cells, but, depending on the regimen (ie, sublethal irradiation), might also provide a competitive advantage to the postnatally transplanted donor cells. There is experimental and clinical evidence for the feasibility of this strategy.74 97 At the present time, most of the other proposed strategies require experimental support.
MATERNAL AND FETAL RISK
Any consideration of fetal therapy must take into consideration maternal and fetal risk. The risks of IUHSCTx can be divided into procedural risks and biological risks for the mother and fetus. The procedural risks are relatively well characterized and can be extrapolated from extensive obstetric experience with chorionic villus sampling (CVS), amniocentesis, and fetal transfusion and blood sampling. The maternal risks from these procedures (ie, infection, hemorrhage, infertility) independent of fetal loss is negligible. The risk of fetal loss or other fetal complications from CVS has been well documented and is less than 1%.98 The procedural risk of IUHSCTx before 14 weeks’ gestation has been previously analyzed and is probably also less than 1% per transplant.2 Therefore, using our current protocol of 3 transplants, we would anticipate a procedural fetal loss rate of no more than 4%. The biologic risks to the mother and fetus include infection with bacterial, fungal, or viral pathogens from the donor cells, fetal GVHD, Rh sensitization for future pregnancies (if the donor cells are Rh-positive and mother and fetus are Rh-negative), and maternal graft-versus-host phenomenon (“autoimmune” disease) if donor lymphocytes cross the placental barrier and survive in the mother.99 Many of these risks can be minimized by using adult sources of donor cells (rather than fetal liver) with careful screening for infectious disease, and scrupulous T-cell depletion. We currently limit the T-cell dose to less than 1 × 105CD3+ cells/kg estimated fetal weight as a precaution against GVHD. The risk of Rh sensitization can be avoided by the use of Rh-negative donor cells, if possible; if not, sensitization can be prevented by administration of Rh-immune globulin. The risk of donor cells crossing the placenta and surviving in the maternal circulation is probably remote, but no data exist. An important concern is whether IUHSCTx would in any way prohibit what is considered current optimal standard of care for a given disease after birth. At the present time there is no rationale to expect that it will, and in fact there is good reason to think that it can potentially facilitate postnatal SCT if tolerance is achieved.
DISEASES THAT MAY CURRENTLY BENEFIT FROM IUHSCTx
It is clear from the above discussion that there are a large number of diseases that might be considered as targets for IUHSCTx. However, it is also clear that each disease must be considered individually and may or may not have favorable enough biology or an adequate rationale for attempting IUHSCTx. Table 2categorizes selected candidate diseases by rationale for IUHSCTx. In contrast to a decade ago, there is now adequate clinical and experimental information available to guide rational clinical application of this approach.
In contrast to postnatal SCT, IUHSCTx strives to create a level of mixed chimerism adequate to ameliorate the clinical manifestations of the disease. Therefore, in consideration of various diseases, two important questions must be asked: (1) What level of engraftment would be adequate to treat a specific disease? and (2) Is there reason to believe a competitive advantage for donor cells is present? From the preceding discussion, it would be unreasonable at the present time to expect a conventional protocol for IUHSCTx to be successful, in the absence of either a selective advantage for donor cells, or the requirement for a very low level of donor cell engraftment to treat the disease. This limits considerably the number of diseases for which IUHSCTx, as currently practiced, can be rationally applied with reasonable expectation of success.
The most biologically favorable target diseases are those that offer a prenatal selective advantage for donor cells. The best examples of diseases in this category are the SCID disorders, particularly the characterized mutations encoding the common cytokine receptor γ chain (X-SCID), or components of it’s signaling pathway (ie, Jak 3 or ZAP-70).100 Based on the available clinical and experimental evidence, it is likely that any member of this group of disorders can be effectively treated by IUHSCTx, using established protocols, with results comparable to the reported results for X-SCID.38 It is important to emphasize that early postnatal T-cell–depleted haploidentical SCT is highly successful for T-cell reconstitution in these patients, but that B-cell function often remains deficient.101 Whether IUHSCTx can improve on the reconstitution achieved by early postnatal transplantation can only be addressed by further clinical trials comparing the two approaches.
Another immunodeficiency disorder in which a selective advantage for normal cells exists is Wiskott-Aldrich syndrome (WAS). Direct evidence of a selective advantage for normal cells is documentation of nonrandom inactivation of the X chromosome in multiple lineages of peripheral blood cells102,103 and early lineage hematopoietic cells104 in carriers of WAS, similar to that seen in the T-cell lineage in carriers of X-SCID.105 A selective advantage for normal progenitors and a proliferative defect in host T cells should provide favorable biology for successful IUHSCTx.
A selective advantage for normal cells would also be expected in diseases in which somatic mosaicism and spontaneous reversion have been documented to occur. In these diseases there is presumably a survival advantage for the spontaneously corrected cells.106 Such correction has been noted in adenosine deaminase deficient (ADA) SCID,107 Fanconi anemia,108 and Bloom’s syndrome,109 the latter two of which are chromosomal breakage syndromes. This experiment of nature shows the potential for selective expansion of a small number or single clone of normal cells with correction of the disease.
Finally, in this category (and possibly the next) are diseases that require very low levels of donor cell engraftment to effect a cure. Therapeutic or near-therapeutic levels of engraftment might be achievable by “standard” protocols of IUHSCTx in diseases such as CGD and hyper IgM syndrome. It has been well documented in animal models that the immune deficit in CGD can be corrected by as few as 5% normal neutrophils.95 In X-linked hyper IgM syndrome, a disease caused by a mutation in the CD40 ligand on T cells,110 carriers have been identified in which the normal gene has been predominantly silenced.111 In these carriers even a few percent of T cells expressing the normal gene can result in normal class switching and IgG production. As discussed above, two recent attempts to treat fetuses affected by CGD with enriched paternal BM have resulted in no detectable engraftment, suggesting that to effectively treat these diseases further optimization of current IUHSCTx protocols will be needed.
DISEASES THAT MAY BENEFIT FROM IUHSCTx IN COMBINATION WITH POSTNATAL STRATEGIES
Although in the absence of a selective advantage only low-level chimerism can be reasonably expected after IUHSCTx, low-level chimerism may carry with it the tremendous advantage of donor-specific transplantation tolerance. This would have the clinical effect of providing a donor without antigenicity after birth. As discussed above, in the absence of immune response, there is increasing evidence that engraftment can be achieved with minimally myeloablative strategies. Particularly for diseases that have been shown to be treatable by stable mixed chimerism, postnatal “booster” transplants could be performed to augment the minimal chimerism achieved in utero with relatively minimal toxicity (Fig 3).

Strategy of combined IUHSCTx and minimally myeloablative same-donor postnatal SCT. This strategy presupposes the use of adult-derived donor cells and is dependent on specific tolerance induction and, presumably, the establishment of at least minimal donor HSC engraftment. Methods to improve the efficiency of tolerance induction such as cotransplantation of donor-derived antigen-presenting cells (APCs) may prove useful in the future. If effective, this strategy could be rationally applied to diseases ameliorated by low or moderate levels of mixed hematopoietic chimerism and, potentially, to any disease effectively treated by postnatal SCT in which a matched sibling donor is not available.
Strategy of combined IUHSCTx and minimally myeloablative same-donor postnatal SCT. This strategy presupposes the use of adult-derived donor cells and is dependent on specific tolerance induction and, presumably, the establishment of at least minimal donor HSC engraftment. Methods to improve the efficiency of tolerance induction such as cotransplantation of donor-derived antigen-presenting cells (APCs) may prove useful in the future. If effective, this strategy could be rationally applied to diseases ameliorated by low or moderate levels of mixed hematopoietic chimerism and, potentially, to any disease effectively treated by postnatal SCT in which a matched sibling donor is not available.
The hemoglobinopathies are the primary candidate diseases for this approach.112 BM transplantation (BMT) is currently the only curative therapy for the hemoglobinopathies. However, in the absence of a matched sibling donor, results have been compromised by treatment-related toxicity,113-116 limiting the option of cellular therapy to the minority of affected patients. The early gestational prenatal diagnosis of the hemoglobinopathies is well established and could potentially be applied to all pregnancies known to be at risk.117 Thus, prenatal strategies warrant consideration.
The prenatal hematopoietic biology of the α- and β-thalassemias and sickle cell disease (SCD), in the context of IUHSCTx, are different and should be considered separately. In the case of α-thalassemia-1, α-globin–dependent hemoglobin production (Hb F) begins at 8 weeks’ gestation.118 By 10 weeks, physiologic evidence of fetal anemia can be observed by ultrasound (placentomegaly). By 12 to 14 weeks, fetal hydrops (high-output cardiac failure) may be observed.119 During this time the fetus develops ineffective erythropoiesis in the fetal liver and abnormal sites of extramedullary hematopoiesis. Therefore, the fetal hematopoietic microenvironment is hypercellular and the fetus is anemic. In contrast, β-globin–dependent hemoglobin production does not occur until after birth. Production of Hb F is normal in β-thalassemia major and SCD during fetal life and, therefore, the fetal microenvironment is relatively normal in its competitive capacity. Ineffective erythropoiesis begins with the switch to adult hemoglobin after birth and may provide a postnatal selective advantage for donor-derived erythropoiesis.
There are experimental data available that support the concept of treatment of thalassemia by the creation of mixed chimerism.120-122 A selective advantage for donor erythropoiesis based on a survival advantage for donor erythroid-committed progenitors due to ineffective host erythropoiesis and/or the relative half-life of mature donor and recipient red blood cells may exist. Clinical observations on mixed chimerism after postnatal BMT for β-thalassemia123,124 and SCD115 support the presence of a selective advantage for donor-derived erythropoiesis. In addition, in β-thalassemia there is evidence that second transplants using minimally myeloablative regimens can augment the presence of minimal chimerism.97
DISEASES THAT ARE UNLIKELY TO BENEFIT FROM IUHSCTx
This category of diseases would include hematologic disorders not cured by postnatal BMT, diseases that have no selective advantage and require near-complete hematopoietic replacement to effect a cure, and diseases that have central nervous system (CNS) manifestations requiring large-scale CNS repopulation for prevention of the clinical disease.
At the present time, most metabolic storage diseases would fall into this category. In some storage diseases there has been no benefit or improvement in the CNS manifestations after BMT (ie, Sanfilippo and Hunter syndromes) and there is no rationale for IUHSCTx.125Those storage diseases that have been corrected by postnatal BMT such as Gaucher’s disease126 or Maroteaux-Lamy syndrome127 (minimal CNS involvement) require higher levels of donor cell engraftment than are currently attainable by IUHSCTx, but could be candidate disorders for prenatal tolerance induction if no matched donor is available. Another group are those in which BMT has corrected the peripheral manifestations of the disease and arrested the neurologic deterioration, but not reversed preexisting neurologic injury, ie, adrenoleukodystrophy and Hurler’s disease.125,128 In these diseases the neurologic injury may begin before birth. The primary question is whether donor HSC-derived microglial elements would populate the CNS providing the necessary metabolic correction within the blood-brain barrier. In a sheep model of Ceroid-Lipofuscinosis, IUHSCTx resulting in levels of donor cell chimerism of approximately 10% in peripheral blood failed to affect the clinical course of the disease.129 Maturation of the blood brain barrier restricts access to the CNS of transplanted cells or the deficient enzyme. Although there is good evidence for origin of CNS glial cells from BM, the timing of their differentiation and migration to the CNS is unclear and probably quite early.130 Thus, even with relatively high levels of systemic engraftment, there may be inadequate enzyme production within the CNS to effect a cure. At the present time, it would seem that this group of diseases is the least biologically attractive group for IUHSCTx. In the future, new approaches such as CNS-directed gene therapy, or cellular therapy131 using CNS “stem cells” or “mesenchymal stem cells” in combination with IUHSCTx, can be envisioned.
ETHICAL CONSIDERATIONS OF THE FETUS AS A PATIENT
Because of the experimental nature of IUHSCTx and considerations unique to fetal therapy in general, it is essential to develop an ethical framework for nondirective counseling of patients and families. Fortunately, the ethical framework for the field of fetal therapy has been relatively well developed.132 The concept of the fetus as a patient, and when a fetus has independent moral status, has been rationally addressed by McCullough and Chervenek.133 They argue that the previable fetus has no independent moral status, and that all arguments to that effect on religious, philosophic, or moral grounds are impossible to bring to closure. Rather, the previable fetus should be considered to have “dependent” moral status. The status of the previable fetus is totally dependent on the mother’s autonomous decisions. It follows that only she can present the fetus as a patient for treatment. However, because of the experimental nature of IUHSCTx, she has no moral obligation to present her fetus for experimentation. Thus, the moral imperative for the investigator is to respect the pivotal status of the mother in the decision to treat her fetus. Implicit in this moral imperative is the necessity for accurate, honest, and nondirective counseling in the informed consent process. A key component of counseling is to assure that the parents understand the options, including postnatal treatment, and potential outcomes. This requires discussion with pediatric bone marrow transplant physicians or immunologists regarding their future child’s disorder and prospects with conventional treatment. It is inappropriate to convince a patient who would otherwise terminate the pregnancy that an unproven, experimental procedure (ie, IUHSCTx) is her best option.
The ethical questions in case selection are also similar in many respects to those with other fetal therapeutic endeavors and can be discussed in the context of benefit and risk to the subjects involved. Determination of risk and benefit may be somewhat arbitrary and may be influenced by a mother’s experience with previous children or family members. The assessment of benefit is complicated by variable phenotypic expression of some genotypes, ie, it may be difficult to predict the severity of some diseases based on known genotype and previous family history. If risks are low, the required benefit can be relatively small and a wider spectrum of candidate diseases could be treated. If, on the other hand, risk is high, then the required expectation of benefit should be high and only a few diseases would be reasonable candidates. Until wider experience is gained with IUHSCTx, this risk-to-benefit analysis will remain imperfect, but at the present time the risks appear small relative to the potential benefits. Therefore, it seems premature to make absolute statements about appropriate candidate diseases for prenatal therapy, although application should be based on rational expectation of success based on a thorough understanding of the field. Further insights into the biology of prenatal transplantation will undoubtedly yield more successful therapeutic strategies based on IUHSCTx in the future.
Supported in part by Public Health Service Grants No. HL 52954 (A.W.F.), HL49042-04 (E.D.Z.), HL48378 (E.D.Z.), DK51427 (E.D.Z.), and HL52955 (E.D.Z.); funds from the Ruth and Tristam C. Colket Jr. Chair in Pediatric Surgery; and by the G. Harold and Leila Y. Mather’s Charitable Foundation.