

Abstract
Histidine-rich glycoprotein (HRG) circulates in plasma at a concentration of 2μM and binds plasminogen, fibrinogen, and thrombospondin. Despite these interactions, the physiologic role of HRG is unknown. Previous studies have shown that mice and humans deficient in HRG have shortened plasma clotting times. To better understand this phenomenon, we examined the effect of HRG on clotting tests. HRG prolongs the activated partial thromboplastin time in a concentration-dependent fashion but has no effect on tissue factor–induced clotting, localizing its effect to the contact pathway. Plasma immunodepleted of HRG exhibits a shortened activated partial thromboplastin time that is restored to baseline with HRG replenishment. To explore how HRG affects the contact pathway, we examined its binding to factors XII, XIIa, XI, and XIa. HRG binds factor XIIa with high affinity, an interaction that is enhanced in the presence of Zn2+, but does not bind factors XII, XI, or XIa. In addition, HRG inhibits autoactivation of factor XII and factor XIIa–mediated activation of factor XI. These results suggest that, by binding to factor XIIa, HRG modulates the intrinsic pathway of coagulation, particularly in the vicinity of a thrombus where platelet release of HRG and Zn2+ will promote this interaction.
Introduction
Despite the capacity of the intrinsic pathway to enhance thrombin generation, patients deficient in factor XII (FXII) do not bleed.1 Even patients with FXI deficiency rarely have hemorrhagic complications, except with surgery or major trauma.2 Because of these observations, it is well accepted that the contact system plays little part in hemostasis; and, by extension, it was also thought to be unimportant for thrombosis. However, a number of recent studies challenge this thinking. First, mice with deficiencies of high- molecular-weight kininogen (HK), bradykinin B2 receptor, FXII, or FXI are protected against injury-induced thrombosis, and an antibody against FXI inhibits thrombus formation in a baboon arteriovenous shunt model.3-7 These observations raise the possibility that the contact pathway contributes to thrombogenesis.8 Second, potential physiologic activators of FXII have been identified, which could initiate the contact system at sites of vascular injury. In addition to glycosaminoglycans and collagen, novel activators include polyphosphates and RNA.9,10 Polyphosphates, which are released from the dense granules of platelets activated at sites of injury, trigger coagulation in a FXII-dependent fashion. Likewise, RNA released from the damaged vessel wall also can activate FXII, and RNAase administration to animals attenuates thrombosis at sites of injury, observations that have sparked a renewed interest in the contact pathway.10 Consequently, it is important to better understand this pathway and how it is regulated.
Histidine-rich glycoprotein (HRG) is an abundant plasma protein whose role is largely unknown.11 HRG circulates at a concentration of approximately 2μM. Its concentration may increase locally because HRG also is found in α-granules of platelets where it can be released on activation.12 HRG is a modular protein composed of 2 NH2-terminal cystatin-like domains, a central histidine-rich region, and a COOH-terminal domain. These domains contribute to binding of Zn2+ and numerous ligands, including plasminogen, fibrinogen, thrombospondin, and heparan sulfate. Because of its multiple interactions, it has been hypothesized that HRG contributes to hemostasis as an accessory or adapter protein.11,13 In support of this concept, HRG-deficient mice exhibit shortened clotting times.14 Furthermore, HRG has been shown to inhibit contact activation of plasma in vitro, presumably because the histidine-rich region of HRG binds to negatively charged surfaces and prevents autoactivation of FXII.15 Modulation of the contact system may impact thrombosis because studies have linked deficiency of HRG in humans with thrombophilia.16 The current study was undertaken to more precisely define the role of HRG in the modulation of coagulation.
Methods
Reagents
Deaminated heparin (molecular weight 6700), biotinamidohexanoic acid hydrazide (biotin-hydrazide), Protease Inhibitor Cocktail for His-Tagged Proteins, and silica (0.5-10 μm particle size) were from Sigma-Aldrich. Dextran sulfate (500 kDa) was from GE Healthcare. Sulfosuccinimidyl-6-biotinamido hexanoate was from Thermo Fisher Scientific. All coagulation factors and corn trypsin inhibitor (CTI) were from Enzyme Research Laboratories. A sheep polyclonal antibody against human HRG, FXII-deficient plasma, and the FXII enzyme immunoassay kit were from Affinity Biologicals. Chromogenic substrates S-2302, S-2366, and S-2251 were from DiaPharma Group, Biophen CS11–22 was from Aniara, and Pefachrome XIIa was supplied by Pentapharm. Human C1 inhibitor was from EMD Chemicals. Recombiplastin and APTT-SP were from Instrumentation Laboratory.
HRG was isolated by a modification of a previously published method.17 Citrated plasma was supplemented with 1μM Val-Phe-Lys-chloromethyl ketone (EMD Chemicals), 0.1% protease inhibitor cocktail, and approximately 50 kallikrein inhibitory units (KIU) aprotinin (American Diagnostica). After centrifugation at 10 000g for 30 minutes at 4°C, any visible lipid layer was removed and solid imidazole was added to 5mM. Plasma was subjected to chromatography on a 50-mL nickel-nitrilotriacetate agarose column (QIAGEN). The column was washed with 5 volumes of 20mM Tris-HCl, 150mM NaCl, pH 7.4 (Tris-buffered saline [TBS]) containing 5mM imidazole. Successive washes with TBS contained 5mM imidazole, 5mM imidazole plus 10mM ϵ-amino caproic acid, 80mM imidazole, and then 100mM imidazole. The ϵ-amino caproic acid was present because plasminogen is a frequent contaminant.18 Aprotinin was added to the 80 and 100mM imidazole wash buffers at 10 KIU/mL. HRG was eluted with TBS containing 250mM imidazole, pH 7.5. The concentration of HRG was determined by absorbance at 280 nm, using a molecular weight of 67 000 and an extinction coefficient of 0.39 mL/mg/cm.19,20 As reported previously, HRG appeared as a closely migrating doublet on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing and reducing conditions (not shown).12 The identity of HRG was confirmed by NH2-terminal sequence analysis (Hospital for Sick Children: Advanced Protein Technology Center Peptide Sequencing Facility, Toronto, ON) and by mass spectrometric analysis of tryptic fragments (Alphalyse Canada).
HRG-deficient plasma was prepared by repeated chromatography over an immobilized polyclonal antibody against HRG. A second sample of plasma was subjected to chromatography on unmodified Sepharose for use as a control. Removal of HRG from plasma was verified by Western blot analysis using the same antibody.
Influence of HRG on FXII autoactivation
FXII autoactivation, stimulated by polyphosphate (Sigma-Aldrich type 65), was evaluated at 23°C in the absence or presence of HRG. A total of 100nM FXII was incubated in 20mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid-OH, 150mM NaCl, pH 7.4 (HBS), containing 2mM CaCl2, 12.5μM ZnCl2, and 100μM polyphosphate (monomer concentration). Samples were mixed in the presence of increasing concentrations of HRG for 15 minutes at 37°C. After addition of 500μM Biophen CS11–22, absorbance was measured at 405 nm in a plate reader (Molecular Devices). Rates of cleavage of the chromogenic substrate were normalized relative to that obtained in the absence of HRG, and plotted against HRG concentration. Data were analyzed with TableCurve 2D Version 4 (Jandel Scientific Software, SPSS) using a rectangular hyperbola equation to determine the concentration of HRG required to produce 50% inhibition of activation (IC50). In similar experiments, 1% APTT-SP, 2 μg/mL dextran sulfate, or 10 μg/mL silica was used in place of polyphosphate. Experiments with silica were performed in the absence or presence of 3μM fibrinogen.
Influence of HRG on FXI activation by FXIIa
The effect of HRG on FXI activation by FXIIa was examined in the presence of APTT-SP and HK. Samples containing 10nM FXIIa, 100nM HK, 120nM FXI, 10% APTT-SP, and 0 to 2μM HRG in HBS containing 2mM CaCl2 and 12.5μM ZnCl2 were incubated at 37°C. After 60 minutes, aliquots were removed and the extent of FXI activation was determined by monitoring FXIa-mediated hydrolysis of 600μM S-2366. Values were normalized relative to that obtained in the absence of HRG and the IC50 was determined.
Influence of HRG on FXII activation by kallikrein
SDS-PAGE was used to monitor the activation of 2.5μM FXII by 100nM kallikrein in a mixture of 2 μg/mL dextran sulfate, 12.5μM CTI, 2mM CaCl2, and 12.5μM ZnCl2 in the absence or presence of HRG. At time points ranging from 0 to 2 hours, 10-μL aliquots containing 2 μg of FXII were removed and subjected to SDS-PAGE under reducing conditions. In the reciprocal experiment, the influence of HRG on prekallikrein activation by FXIIa was monitored in the absence or presence of 600nM HK. After incubation of 120nM prekallikrein with 2nM FXIIa in TBS containing 2mM CaCl2, 12.5μM ZnCl2, and 0.5 μg/mL dextran sulfate for 2 minutes, kallikrein generation was monitored using S-2302.
Biotinylation
Biotin labeling was carried out for 1.5 hours at 23°C. HRG was dialyzed against 0.1M phosphate buffer, pH 7.5, and incubated with a 10-fold molar excess of sulfosuccinimidyl-6-biotinamido hexanoate. To stop the reaction, 16μM glycine, pH 8.0, was added and free biotin was removed by passage over a PD-10 column (GE Healthcare) equilibrated with HBS. Biotin-hydrazide was reacted with deaminated heparin at a 10:1 molar ratio. The sample was dialyzed against water and the biotin-labeled heparin was lyophilized and its concentration determined by mass. FXIIa was biotin-labeled at its active site by reaction with biotin-Phe-Pro-Arg chloromethyl ketone (b-FPR; Haematological Technologies).
Surface plasmon resonance
Biomolecular interaction analysis was performed on the Biacore T100 biosensor system (BIAcore). Biotinylated-HRG was adsorbed to 800 response units (RU) onto a Series S sensor chip SA in HBS containing 0.05% Tween 20 (HBS-T) at a flow rate of 5 μL/min. An unmodified flow cell served as control. First, using dual injection mode, 10nM FXIIa was injected together with 0 to 20μM ZnCl2 at 10 μL/min followed by a second injection of ZnCl2 alone. A second dissociation phase was monitored with ZnCl2-free buffer, and then flow cells were regenerated with 250mM imidazole and 5mM ethylenediaminetetraacetic acid. RU values were obtained from the instrument software, and association and dissociation rate constants and RUmax values were used for determination of the dissociation constant (Kd). Second, to determine the affinity of Zn2+ for HRG, ZnCl2, in concentrations from 0 to 10μM, was injected at 30 μL/min over flow cells containing immobilized HRG and RU values were determined. Next, to confirm the Kd values of FXIIa for HRG, b-HRG was immobilized to 50 to 100 RU. FXIIa (0-256nM) was injected at a flow rate of 45 μL/min for 10 minutes in the absence or presence of 12.5μM ZnCl2, followed by buffer alone. Kd values were determined from equilibrium values or calculated from rate constants using a 1:1 Langmuir analysis by the instrument software, and χ2 values were calculated to assess closeness of fit.
Binding affinity also was determined on a BIAcore 1000 using immobilized b-FPR-FXIIa and soluble HRG. b-FPR-FXIIa (800-900 RU) was absorbed to a BIAcore SA streptavidin chip at a flow rate of 5 μL/min at 25°C. HRG, diluted in HBS-T containing 12.5μM ZnCl2 and 2mM CaCl2, or 2mM ethylenediaminetetraacetic acid, at pH 7.4, was injected at 35 μL/min for approximately 4 minutes, followed by a 4-minute injection of HBS-T. Increasing concentrations of analyte (0-2000nM) were injected over the flow cell, as well as over a blank streptavidin-coated flow cell. Sensorgrams were analyzed with BIAevaluation software Version 3.2 (BIAcore), or Scrubber2 software Version 2.0a (BioLogic Software Pty). A competition experiment was conducted to examine the effect of FXII or FXIIa on the interaction of HRG with heparin. Samples containing 250nM HRG were injected over the biotin-heparin modified chip (200 RU) in the presence of 0 to 400nM FXII or FXIIa and RU values were determined.
Clotting assays
In a multiwell plate, 90-μL aliquots of citrated, normal pooled human plasma were incubated with 0 to 5μM HRG for 10 minutes at 37°C before addition of 10 μL of 260mM CaCl2 and 10 μL of APTT-SP. The plate was then read at 405 nm for 30 minutes at 37°C, and the time to reach half-maximal absorbance was determined by Soft Max Pro Version 5.4 (clot time). To examine tissue factor-mediated clotting, Recombiplastin, at one-tenth dilution, was substituted for APTT-SP. Additional assays were performed in HRG-deficient plasma that was first dialyzed against TBS to remove citrate. In these assays, clotting was initiated with 8nM FXIIa, 1.5nM FIXa, or 2nM FXIa, which was added to 100 μL of plasma together with 2mM CaCl2, 15μM ZnCl2, and 0 to 5μM HRG in a total volume of 160 μL. FXIIa-mediated clotting of normal plasma was also assessed in the absence or presence of 3.5 mg/mL sheep anti-HRG antibody or control sheep IgG (Sigma-Aldrich).
Determination of plasma HRG concentration
The concentration of HRG in normal plasma was determined to be 1.6μM by surface plasmon resonance (SPR; supplemental Methods, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Influence of HRG on inhibition of FXIIa by C1 inhibitor or CTI
This information is contained in supplemental Methods.
Detection of HRG-FXIIa complexes in plasma
This information is contained in supplemental Methods.
Results
Effect of HRG on plasma clotting times
HRG was added to normal citrated plasma, and clotting was initiated with tissue factor or APTT-SP to examine the effect of HRG on the extrinsic and intrinsic pathways, respectively. HRG had little effect on tissue factor–induced clotting, where the clotting times in the absence and presence of 3.2μM HRG were 79.7 ± 2.1 and 83.8 ± 4.9 seconds, respectively (P = .47). In contrast, the activated partial thromboplastin time of control plasma (248 ± 4 seconds) was 4-fold longer in the presence of 2μM HRG (1057 ± 127 seconds; P = .008). These data suggest that HRG affects clotting through the intrinsic pathway and does not influence the extrinsic pathway.
To examine this more closely, the effect of added HRG on clotting of HRG-deficient plasma was examined. To minimize the impact of plasma processing, control plasma was subjected to chromatography on unmodified Sepharose. In the APTT assay, HRG-deficient plasma clotted 2-fold more rapidly than control plasma (216 ± 24.1 and 426 ± 10.1 seconds, respectively; P < .001). These data confirm that HRG attenuates the contact pathway of clotting. When HRG was added back to the deficient plasma, a dose-dependent and saturable increase in clotting time was observed (Figure 1).

Effect of HRG on the activated partial thromboplastin time in HRG-deficient plasma. HRG-deficient plasma was incubated with 0 to 6μM HRG for 10 minutes at 37°C. Clotting was initiated by the addition of CaCl2 to 26mM and one-tenth volume of APTT-SP. Absorbance was monitored at 405 nm, and the time to half-maximal value was defined as the clot time. Data are the mean ± SE of 3 determinations. The control was plasma subjected to chromatography on unmodified Sepharose.
Effect of HRG on the activated partial thromboplastin time in HRG-deficient plasma. HRG-deficient plasma was incubated with 0 to 6μM HRG for 10 minutes at 37°C. Clotting was initiated by the addition of CaCl2 to 26mM and one-tenth volume of APTT-SP. Absorbance was monitored at 405 nm, and the time to half-maximal value was defined as the clot time. Data are the mean ± SE of 3 determinations. The control was plasma subjected to chromatography on unmodified Sepharose.
To identify where HRG was affecting the contact system, we next examined its effect on clotting of HRG-deficient plasma initiated by FXIIa, FXIa, or FIXa. HRG prolonged the FXIIa-mediated clotting time by 81% and 102% at 2.5 and 5μM, respectively (Figure 2). In contrast, HRG had little effect on FXIa- or FIXa-mediated clotting times. These results suggest that HRG prolongation of the APTT reflects an effect on FXIIa. To confirm this, we measured the APTT in the absence or presence of the HRG-directed antibody. Antibody addition shortened the clotting time from 358 ± 55 seconds to 140 ± 19 seconds (Figure 3; P = .02), whereas a nonspecific antibody had no effect.

Effect of HRG on clotting induced by FIXa, FXIa, or FXIIa in HRG-deficient plasma. HRG-deficient plasma was dialyzed to remove citrate and supplemented with 2mM CaCl2, 12.5μM ZnCl2, and increasing concentrations of HRG. Clotting at 37°C was initiated by the addition of 1.5nM FIXa (■), 0.5nM FXIa ( ), or 5nM FXIIa (□) and monitored by turbidity. Clotting times are plotted versus HRG concentration. Data are mean ± SE of 3 determinations.
), or 5nM FXIIa (□) and monitored by turbidity. Clotting times are plotted versus HRG concentration. Data are mean ± SE of 3 determinations.
Effect of HRG on clotting induced by FIXa, FXIa, or FXIIa in HRG-deficient plasma. HRG-deficient plasma was dialyzed to remove citrate and supplemented with 2mM CaCl2, 12.5μM ZnCl2, and increasing concentrations of HRG. Clotting at 37°C was initiated by the addition of 1.5nM FIXa (■), 0.5nM FXIa (), or 5nM FXIIa (□) and monitored by turbidity. Clotting times are plotted versus HRG concentration. Data are mean ± SE of 3 determinations.

Effect of anti-HRG antibody on FXIIa-mediated clotting of plasma. Normal plasma was incubated with 5 mg/mL control or anti-HRG sheep IgG for 50 minutes. Aliquots were made to 18mM CaCl2 and 5nM FXIIa, and absorbance was monitored at 405 nm. Clot times were determined in triplicate and are plotted as mean ± SE.
Effect of anti-HRG antibody on FXIIa-mediated clotting of plasma. Normal plasma was incubated with 5 mg/mL control or anti-HRG sheep IgG for 50 minutes. Aliquots were made to 18mM CaCl2 and 5nM FXIIa, and absorbance was monitored at 405 nm. Clot times were determined in triplicate and are plotted as mean ± SE.
Binding of Zn2+ to HRG
Plasma-based assays were performed largely in the absence of Zn2+ because citrate was used as the anticoagulant. However, because HRG is a Zn2+ binding protein and Zn2+ modulates its properties, Zn2+ was included in our characterization of the molecular interactions of HRG with contact pathway proteins. To identify the optimal concentration of Zn2+ for these studies, we first determined its affinity for HRG using SPR. Biotin-HRG was adsorbed to a streptavidin chip, and increasing concentrations of ZnCl2 were injected with buffer regeneration between runs. RU values increased in a dose-dependent fashion reflecting Zn2+ binding (Figure 4). Analyses by kinetic and equilibrium parameters yielded Kd values of Zn2+ for HRG of 225 and 281nM, respectively (χ2 ≤ 1.2). Because saturable binding was observed at concentrations greater than 5μM Zn2+, subsequent experiments were performed at 10 to 20μM Zn2+, which coincides with the plasma Zn2+ concentration.21
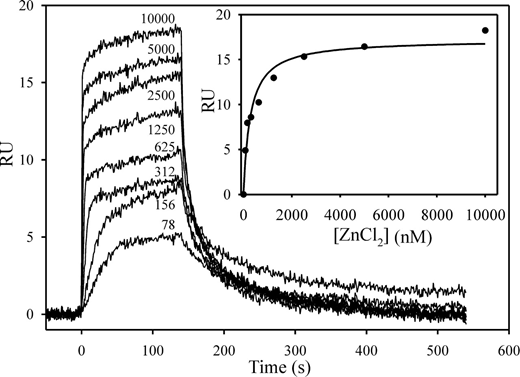
Binding of Zn2+ to HRG monitored by SPR. Biotin-HRG was immobilized on a streptavidin-modified chip to 800 RU. ZnCl2 (0-10 000nM) was injected over the flow cell for 120 seconds, and then buffer was injected. ZnCl2 concentrations in nanomolar are noted beside each tracing. (Inset) A plot of the RU values at equilibrium versus the concentration of ZnCl2. The data were analyzed by rectangular hyperbola equation (line).
Binding of Zn2+ to HRG monitored by SPR. Biotin-HRG was immobilized on a streptavidin-modified chip to 800 RU. ZnCl2 (0-10 000nM) was injected over the flow cell for 120 seconds, and then buffer was injected. ZnCl2 concentrations in nanomolar are noted beside each tracing. (Inset) A plot of the RU values at equilibrium versus the concentration of ZnCl2. The data were analyzed by rectangular hyperbola equation (line).
Effect of HRG on the autoactivation of FXII
FXII (100nM) was incubated with 0 to 2μM HRG in the presence of 12.5μM ZnCl2, 2mM CaCl2, and 100μM polyphosphate. After 10 minutes of incubation, FXIIa activity was monitored by chromogenic assay. Rates were plotted versus HRG concentration and analyzed by nonlinear regression (Figure 5A). HRG inhibited FXII autoactivation by more than 95% and inhibition was concentration-dependent with an IC50 of 115nM. HRG also inhibited autoactivation mediated by APTT-SP, silica, or dextran sulfate in a similar fashion (not shown). Although this activity could reflect the capacity of HRG to bind and neutralize the activator, further investigation indicated that HRG binds dextran sulfate but not silica, the active agent in APTT-SP (not shown). These data suggest that HRG does not attenuate coagulation simply by compromising the contact activator.

Influence of HRG on FXII autoactivation and FXI activation. (A) FXII was incubated with 100μM polyphosphate for 15 minutes in HBS buffer containing 2mM CaCl2 and 12.5μM ZnCl2. After 10 minutes, aliquots were removed and FXIIa generation was determined by monitoring the hydrolysis of 500μM Biophen CS11–22. The data points represent the mean ± SE of 2 separate experiments. Data were analyzed by nonlinear regression (line) to determine the IC50. (B) To examine the influence of HRG on FXI activation, 10nM FXIIa was incubated with 0 to 1μM HRG, 100nM HK, 10% APTT-SP, and 120nM FXI in HBS buffer containing 12.5μM ZnCl2 and 2mM CaCl2. Aliquots were removed, and FXIa generation was determined by monitoring the hydrolysis of 600μM S-2366. Rates of chromogenic substrate cleavage in the presence of HRG were normalized relative to that obtained in its absence. The data were fit to a rectangular hyperbola equation (line) to determine the IC50. The data points represent the mean ± SE of 2 experiments.
Influence of HRG on FXII autoactivation and FXI activation. (A) FXII was incubated with 100μM polyphosphate for 15 minutes in HBS buffer containing 2mM CaCl2 and 12.5μM ZnCl2. After 10 minutes, aliquots were removed and FXIIa generation was determined by monitoring the hydrolysis of 500μM Biophen CS11–22. The data points represent the mean ± SE of 2 separate experiments. Data were analyzed by nonlinear regression (line) to determine the IC50. (B) To examine the influence of HRG on FXI activation, 10nM FXIIa was incubated with 0 to 1μM HRG, 100nM HK, 10% APTT-SP, and 120nM FXI in HBS buffer containing 12.5μM ZnCl2 and 2mM CaCl2. Aliquots were removed, and FXIa generation was determined by monitoring the hydrolysis of 600μM S-2366. Rates of chromogenic substrate cleavage in the presence of HRG were normalized relative to that obtained in its absence. The data were fit to a rectangular hyperbola equation (line) to determine the IC50. The data points represent the mean ± SE of 2 experiments.
To begin to investigate the mechanism by which HRG inhibits FXII activation, control experiments were first performed to determine whether HRG binds to the active site of FXIIa in the presence of Zn2+. HRG had variable effects on FXIIa chromogenic activity, depending on the substrate (not shown). With S-2222, no effect of HRG was observed. In contrast, with Pefachrome XIIa, 2μM HRG promoted FXIIa activity by 35%, raising the possibility that HRG binds FXIIa and allosterically modifies its activity. The observation that HRG did not decrease FXIIa chromogenic activity makes it unlikely that HRG obscures the active site of FXIIa. To verify this concept, the effect of HRG on FXIIa inhibition by C1 inhibitor or CTI was examined in the presence of Zn2+. The second-order rate constant for FXIIa inhibition by C1 inhibitor in the absence of 5μM HRG was similar to that in its presence (1.3 ± 0.05 × 105 and 1.1 ± 0.07 × 105M−1min−1, respectively). Likewise, HRG also had minimal effect on the inhibition of FXIIa by CTI, with second-order rate constants of 4.5 ± 0.5 × 106 and 2.9 ± 1.29 × 106M−1min−1 in the absence or presence of 5μM HRG, respectively. These findings suggest that HRG does not interact directly with the active site of FXIIa nor does it sterically hinder access of macromolecular inhibitors to the active site of FXIIa.
Effect of HRG on FXIIa-mediated activation of FXI
Additional intrinsic pathway reactions were examined to reveal whether the effect of HRG was specific for FXII autoactivation. Thus, FXIIa-mediated activation of FXI in the presence of HK was assessed in the absence or presence of HRG. As with autoactivation, HRG caused dose-dependent inhibition of FXIIa-mediated activation of FXI in the presence of Zn2+ (Figure 5B). Activation was reduced by more than 95% with an IC50 of 287nM.
Effect of HRG on the kallikrein system
Because prekallikrein and FXIIa comprise an autolytic feedback loop, reactions involving these factors in their zymogen or active forms were examined. The effect of HRG on the activation of FXII by kallikrein in the presence of Zn2+ was examined first. To avoid autoactivation of FXII by FXIIa, CTI was added to inhibit any FXIIa that was formed and SDS-PAGE analysis was used to monitor FXII activation. In the presence of 2μM HRG, activation of FXII by kallikrein was reduced by 50% (not shown). These results demonstrate that HRG has only a modest inhibitory effect on kallikrein activity. In the reciprocal experiment, the effect of HRG on the activation of PK by FXIIa in the presence of Zn2+ was investigated in the absence or presence of HK. In the absence of cofactor HK, HRG inhibited PK activation by 75% with an IC50 of 160nM. When HK was present, the magnitude of inhibition was similar, but the IC50 value was 23-fold higher. These findings suggest that HRG inhibits FXIIa-mediated activation of PK, an effect that is attenuated in the presence of HK. Taken together, the results of these experiments suggest that the major effect of HRG is on FXIIa activity and not FXII activation. This concept was tested in binding experiments.
Binding of HRG to components of the intrinsic pathway
The affinity of HRG for FXIIa was determined by SPR where immobilized b-HRG was titrated with increasing concentrations of FXIIa in the absence or presence of 12.5μM Zn2+ (Figure 6A-B). Zn2+ had little effect on association but considerably slowed dissociation. Analysis of the rates indicated a Kd of HRG for FXIIa of 21nM (χ2 = 2.6) in the presence of ethylenediaminetetraacetic acid and 7.5pM (χ2 = 7.0) in the presence of Zn2+. Binding of HRG to FXIIa was reversible because the interaction was abrogated by injection of imidazole. To investigate the Zn2+ dependence of binding, FXIIa was injected in the presence of increasing concentrations of ZnCl2 (Figure 6C). With increasing concentrations of Zn2+, dissociation of FXIIa gradually slowed. When Zn2+ was removed, samples originally containing Zn2+ dissociated more rapidly. Kinetic analyses of the rates indicated a dose-dependent decrease in the Kd, from 1.6nM without Zn2+ to 9pM in the presence of 20μM Zn2+ (χ2 = 3.7). These results demonstrate that Zn2+ increases the affinity of HRG for FXIIa by 1000-fold and this effect is exerted on the dissociation phase. Inspection of the Zn2+ dependence indicated that the half-maximal effect occurred at 140nM Zn2+, comparable with the Kd of Zn2+ for HRG of 250nM, as determined directly.

Effect of Zn2+ on binding of FXIIa to HRG monitored by SPR. Biotin-HRG was adsorbed onto a streptavidin-coated sensor chip to 50 RU. Increasing concentrations of FXIIa (0-256nM) were injected at a flow rate of 45 μL/min in the presence of 2mM ethylenediaminetetraacetic acid (A) or 12.5μM ZnCl2 (B). FXIIa concentrations are indicated adjacent to each tracing, with the exception of 0.5 and 0.25nM, which were omitted for clarity. FXIIa association to the chip surface was monitored for 8 minutes followed by injection of HBS to monitor dissociation. RU values were corrected for background and plotted versus time. (C) b-HRG (800 RU) was immobilized on a streptavidin chip, and 10nM FXIIa was injected in the presence of 0 to 20μM ZnCl2 for 200 seconds (a), followed by Zn2+-containing buffer lacking FXIIa for 600 seconds (b). Dissociation was then observed in buffer lacking Zn2+ for 400 seconds (c). The micromolar concentrations of ZnCl2 are noted on each tracing.
Effect of Zn2+ on binding of FXIIa to HRG monitored by SPR. Biotin-HRG was adsorbed onto a streptavidin-coated sensor chip to 50 RU. Increasing concentrations of FXIIa (0-256nM) were injected at a flow rate of 45 μL/min in the presence of 2mM ethylenediaminetetraacetic acid (A) or 12.5μM ZnCl2 (B). FXIIa concentrations are indicated adjacent to each tracing, with the exception of 0.5 and 0.25nM, which were omitted for clarity. FXIIa association to the chip surface was monitored for 8 minutes followed by injection of HBS to monitor dissociation. RU values were corrected for background and plotted versus time. (C) b-HRG (800 RU) was immobilized on a streptavidin chip, and 10nM FXIIa was injected in the presence of 0 to 20μM ZnCl2 for 200 seconds (a), followed by Zn2+-containing buffer lacking FXIIa for 600 seconds (b). Dissociation was then observed in buffer lacking Zn2+ for 400 seconds (c). The micromolar concentrations of ZnCl2 are noted on each tracing.
Because of the extremely high affinity of HRG for FXIIa in the presence of Zn2+, the interaction of these 2 proteins was reassessed in a reciprocal experiment. In this experiment, FXIIa, labeled at its active site with b-FPRck, was bound to the SA chip. Using HRG as the analyte, Kd values of 450 ± 40nM and 376 ± 68pM were obtained in the absence or presence of Zn2+, respectively (not shown). Retention of the high-affinity interaction in the reverse binding experiment verifies the initial observation of high-affinity FXIIa binding to HRG in the presence of Zn2+. Further SPR analyses indicated no interaction of HRG with zymogen FXII or biotin-FPR-FXIIaβ (not shown).
To ensure that modification or adsorption of either protein to the SA chip did not lead to spurious results, an additional control was performed in which both proteins were unmodified. SPR experiments indicated that HRG binds heparin with a Kd value of 2nM in the presence of Zn2+, whereas FXII and FXIIa do not bind heparin (not shown). Therefore, biotin-heparin was adsorbed to an SA chip, and binding of HRG was examined in the absence or presence of FXIIa. Injection of HRG in the presence of Zn2+ indicated high-affinity binding to immobilized heparin (Figure 7). In the presence of increasing concentrations of FXIIa, binding of HRG to heparin was reduced in a dose-dependent fashion. At 100nM FXIIa, binding of HRG to heparin was completely inhibited. This result confirms the interaction of HRG with FXIIa and suggests that heparin and FXIIa share the same binding site on HRG. In contrast, zymogen FXII had no effect on the binding of HRG to heparin (not shown). In additional SPR experiments, it was observed that neither FXIa nor FXI binds to immobilized HRG, whereas kallikrein binds HRG with a Kd value of 340nM (not shown).
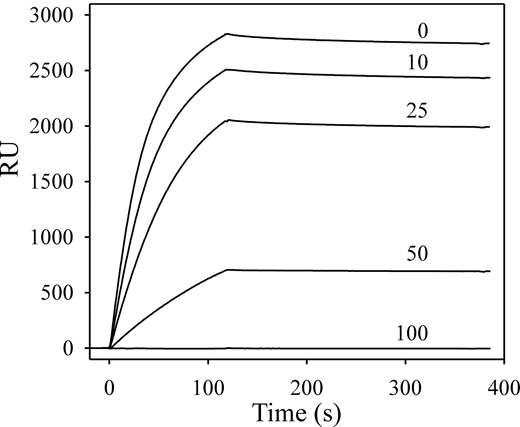
Effect of FXIIa on HRG binding to heparin. Biotin-heparin was adsorbed onto a streptavidin-coated sensor chip to approximately 250 RU. HRG (250nM) was injected in the presence of 10μM ZnCl2, and the association to the chip surface was monitored for 110 seconds before injection of buffer to monitor dissociation. After regenerating the chip, subsequent injections contained 250nM HRG in the presence of 10, 25, 50, or 100nM FXIIa. The binding RU were corrected for background and plotted versus the time course for individual injections. The FXIIa concentrations are indicated above each tracing.
Effect of FXIIa on HRG binding to heparin. Biotin-heparin was adsorbed onto a streptavidin-coated sensor chip to approximately 250 RU. HRG (250nM) was injected in the presence of 10μM ZnCl2, and the association to the chip surface was monitored for 110 seconds before injection of buffer to monitor dissociation. After regenerating the chip, subsequent injections contained 250nM HRG in the presence of 10, 25, 50, or 100nM FXIIa. The binding RU were corrected for background and plotted versus the time course for individual injections. The FXIIa concentrations are indicated above each tracing.
Modulation of HRG activity by fibrinogen
It is apparent that the concentration of HRG required to modulate plasma clotting assays is higher than expected, given that the Kd for FXIIa is 21nM in buffer systems without Zn2+. One potential explanation is that the interaction of HRG with other plasma proteins attenuates its effect on FXIIa. To explore this, the effect of fibrinogen on HRG modulation of FXII autoactivation was examined. Although polyphosphate-mediated autoactivation of FXII was inhibited by 50% at 350nM HRG, the IC50 increased 5-fold to 2000nM in the presence of 3μM fibrinogen (not shown). Thus, considering the number of plasma proteins that bind HRG,22 it is likely that higher concentrations of HRG are required to attenuate FXIIa activity in plasma than predicted solely by its affinity for FXIIa determined in a purified system.
HRG-FXIIa complexes in plasma
An enzyme immunoassay was used to assess the formation of HRG-FXIIa complexes in plasma activated with APTT-SP. Activated plasma yielded an FXIIa-HRG complex concentration of 5 μg/mL, whereas no complexes were detected in unactivated normal plasma or FXII-deficient plasma. The concentration of FXIIa-HRG complexes detected in plasma after APTT-SP addition represents 10% to 20% of the normal plasma concentration of FXII.
Discussion
Emerging evidence suggests that the intrinsic pathway of coagulation contributes to thrombosis at sites of vascular injury.23,24 This has prompted the development of novel anticoagulants that target this pathway.4,25 The success of such an approach depends on an understanding of how the intrinsic pathway is regulated. Because the intrinsic pathway is initiated by the activation of FXII, an understanding of FXIIa regulation is particularly important. The current study was prompted by our initial observation that HRG addition to plasma prolongs the activated partial thromboplastin time but has no effect on the prothrombin time. In accordance with this observation, HRG attenuated clotting initiated by FXIIa, but not by FXIa or FIXa. We showed that HRG binds FXIIa with high affinity and inhibits its capacity to autoactivate FXII or to activate FXI. We also detected HRG-FXIIa complexes in contact-activated plasma. These findings raise the possibility that HRG modulates the intrinsic pathway of coagulation. In support of this concept, immunodepletion of HRG from plasma or addition of an HRG-directed antibody resulted in a shorter activated partial thromboplastin time, whereas HRG supplementation of HRG-deficient plasma prolonged this clotting time.
The available data suggest a mechanism by which HRG and FXIIa interact. Because Zn2+ heightens the affinity of FXIIa for HRG by more than 3 orders of magnitude, the histidine-rich region may mediate this interaction. However, the Zn2+-dependent interaction of heparan sulfate with HRG is mediated by its NH2-terminal cystatin domains and not its histidine-rich domain, suggesting that Zn2+ binding to the histidine-rich domain modulates other domains in HRG.26-29 Because heparin disrupts the interaction of HRG with FXIIa, it is possible that FXIIa also binds to the cystatin domains and that the affinity of this interaction is heightened by Zn2+ binding to the histidine-rich domain. With respect to FXIIa, the catalytic domain of FXIIa can be excluded because β-FXIIa does not bind HRG. This leaves the heavy chain of FXII as the binding site for HRG. The conformational change30 in FXII zymogen evoked by Zn2+ is insufficient to promote HRG binding. Therefore, the HRG binding site must be cryptic on FXII and only exposed when it is converted to FXIIa. This is analogous to the interaction with fibronectin and a monoclonal antibody against the kringle domain of FXII; these proteins bind FXIIa, but not zymogen FXII.31,32 Chromogenic assays indicate that HRG binds to a site distinct from the active site of FXIIa. Furthermore, HRG does not impair the capacity of either CTI or C1 inhibitor to inhibit FXIIa. Therefore, it is likely that HRG binds to an exosite on FXIIa that mediates FXIIa binding to macromolecular substrates.
The specificity and avidity of HRG binding to FXIIa and the inhibitory effects of HRG on FXIIa activity imply a physiologic role for the interaction. Current thinking is that C1 inhibitor is the physiologic inhibitor of FXIIa, although the second-order rate constant suggests that inhibition is slow.33,34 Given the high affinity of HRG for FXIIa and the abundance of HRG in plasma, HRG is more likely to bind FXIIa than is C1 inhibitor. The affinity of FXIIa for HRG is increased in the presence of Zn2+. Although Zn2+ also binds and modulates FXIIa activity,30,35 the effect of HRG is not Zn2+-dependent because HRG binds FXIIa and modulates clotting even in the absence of Zn2+. However, FXIIa will bind HRG even more avidly at sites of vascular injury where platelet activation leads to release of Zn2+ from α-granules.36 The HRG-FXIIa interaction would serve to attenuate FXIIa-mediated activation of the contact pathway, thereby limiting the procoagulant stimulus. Although it has not been demonstrated, it is possible that dissociation of FXIIa from HRG restores its proteolytic activity, raising the possibility that under certain conditions, FXIIa inhibition may be reversible. This could occur in response to a change in the local pH or Zn2+ concentration,37 or possibly through alternate ligand binding to HRG. Despite the potential for further complexity, our data suggest that HRG modulates FXIIa-mediated initiation of the contact pathway, thereby limiting systemic FXIIa activation and localizing the procoagulant response.
HRG is proposed to play a role in numerous processes, including angiogenesis, apoptosis, and immune regulation.11,22 In a knockout model, mice lacking HRG exhibited a procoagulant phenotype, although involvement of the contact pathway was not examined.14 Furthermore, HRG deficiency in humans has been associated with a thrombotic phenotype, at least in some individuals.38 The current description of modulation of the contact system reveals a potential new function for HRG. In support of this concept, a genome wide association study aimed at investigating factors that influence the activated partial thromboplastin time identified highly associated polymorphisms in the genes for FXII, HK, and HRG.39 The means by which HRG modulates the activated partial thromboplastin time may depend on local conditions. In the absence of vascular injury, HRG is unlikely to affect hemostasis because it does not bind FXII zymogen and because free Zn2+ levels in plasma are less than 1μM.21 When FXII is activated at sites of injury and local Zn2+ levels increase in response to platelet activation, HRG can bind FXIIa with high affinity. HRG is a potent inhibitor of FXIIa procoagulant activity, and its plasma levels are relatively high and do not fluctuate widely, except in pathologic states because HRG is a negative acute phase protein.40 Therefore, rather than acting as a dynamic inhibitor of FXIIa, HRG is more likely to serve as a reservoir for FXIIa formed in situ or for FXIIa that has escaped from the site of injury. In this way, HRG may compensate for the low rate of FXIIa inhibition by C1 inhibitor. Because HRG binds fibrin,41 it is conceivable that HRG is retained within the fibrin clot where it is poised to modulate FXIIa-mediated activation of coagulation. Thus, in addition to its proposed role in fibrinolysis,11,22 HRG may also serve as an effector of coagulation. The homology of HRG with HK, a key modulator of the contact system, provides further support for this proposal.42 Both proteins have cystatin-like and histidine-rich domains and bind glycosaminoglycans, Zn2+, and various enzymatic forms of the contact factors.43 However, HK promotes activation of contact factors and protects FXIa and kallikrein from inhibition by antithrombin, whereas HRG inhibits activation of FXII and has no effect on FXIIa inhibition. Thus, HRG may serve to offset the action of HK and limit propagation of the contact system. In conclusion, with emerging evidence for the role of the contact system in the pathogenesis of thrombosis at sites of vascular injury, our data suggest that HRG may be an important regulator of thrombogenesis.
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported in part by the Canadian Institutes of Health Research (grants MOP 3992, MOP 102735, and CTP 79846), the Heart and Stroke Foundation of Ontario (grants T4729 and T4730), the Heart and Stroke Foundation of Canada (Novel and Exploratory Research Fund grant), and the Ontario Research and Development Challenge Fund. J.L.M. was supported by the Heart and Stroke Foundation of Ontario (Graduate Student Award). J.I.W. holds the Heart and Stroke Foundation of Ontario/J. Fraser Mustard Endowed Chair in Cardiovascular Research and Canada Research Chair (Tier 1) in Thrombosis.
Authorship
Contribution: J.L.M. designed and performed research, analyzed and interpreted data, and wrote the manuscript; A.R.S. designed research, contributed vital new reagents, performed research, and analyzed and interpreted data; J.W.Y., B.A.L., and T.T.V. performed research and analyzed and interpreted data; and J.C.F. and J.I.W. designed research, analyzed and interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeffrey I. Weitz, Thrombosis and Atherosclerosis Research Institute, 237 Barton St East, Hamilton, ON L8L 2X2, Canada; e-mail: weitzj@taari.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal