

Abstract
Allogeneic hematopoietic stem cell transplantation (HSCT) can eradicate chemorefractory leukemia through the graft-versus-leukemia (GVL) activity of donor T cells. However, the clinical success of allo-HSCT is limited by the graft-versus-host disease (GVHD) activity of donor T cells. We have reported previously that donor bone marrow precursors of plasmacytoid dendritic cells (pre-pDCs) can activate donor T cells toward T-helper 1 immune polarization in murine allogeneic HSCT. To optimize the GVL activity of these activated donor T cells and limit their graft versus host activity, we engineered the cellular constituents of an allogeneic hematopoietic stem cell graft with highly purified hematopoietic stem cells, T cells, and pre-pDCs and studied their GVL and GVHD activities in a murine model of allogeneic HSCT. Transplanted donor pre-pDCs expanded in vivo for 2 weeks after transplant, and they markedly augmented the activation and GVL activity of donor T cells while attenuating their GVHD activity, leading to an improved therapeutic index. Bidirectional signaling between donor T cells and donor pDCs with IFN-γ synthesis by donor T cells inducing indoleamine 2,3-dioxygenase synthesis by donor pDCs limited GVHD by altering the balance between donor T-reg and inflammatory T cells. Manipulating the content of donor DC precursors in allogeneic HSCT is a novel method to optimize the balance between GVL and GVHD.
Introduction
Donor T cells are responsible for both GVHD and GVL reactions after allogeneic HSCT. The activation status of T cells is modulated by dendritic cells (DCs), the most potent and professional antigen-presenting cells (APCs).1,2 Both host and donor DCs have been shown to play critical roles in regulating GVHD and GVL effects after MHC-mismatched HSCT.3-7 GVHD can be initiated by residual APCs that directly present host antigen (Ag) to donor T cells,5 whereas GVHD intensity can be modulated by donor APCs that present host Ag to donor T cells via indirect antigen presentation.1,3,8 However, despite extensive investigations of the role of host DCs on GVHD pathophysiology, much less is known about the mechanisms by which donor APCs activate and regulate donor T cells. A previous study by MacDonald et al9 demonstrated that depleting CD11c+ donor conventional DCs (CDCs) reduced the severity of GVHD in mice. The same group then demonstrated that conventional donor cDCs isolated from the spleen are the most effective population in presenting alloantigen and stimulating naive donor T-cell responses early post–bone marrow transplantation (BMT).3 Recently, using 2 allogeneic murine BMT models (C57BL/6→B10.BR and C3H→C57BL/6), we showed that addition of donor bone marrow cells enriched for pre-pDCs to a graft composed of purified HSC and T cells significantly improved long-term leukemia-free survival without increasing GVHD compared with recipients of donor HSC and T cells.10 Of note, higher numbers of IFN-γ–producing donor T cells were seen among recipients of pDCs.10 The aim of the present work was to further define the mechanism by which donor pre-pDCs modulate the alloreactivity of donor T cells. Based on the marked up-regulation of IFN-γ in donor T cells cotransplanted with bone marrow enriched for pre-pDCs,10,11 we hypothesized that IFN-γ–responsive genes in donor pre-pDCs might be involved in their immunomodulatory activity.
Using highly purified populations of donor pre-pDCs, we observed that IFN-γ signaling by donor T cells to donor pre-pDCs led to increased indoleamine-2,3-dioxygenase (IDO) expression in donor pDCs and that IDO production by donor pDCs suppressed the GVHD activity of donor T cells and changed the balance between regulatory and inflammatory donor T cells. These data support a new paradigm for immune regulation in allogeneic HSCT in which donor DCs first activate donor T cells and then subsequently limit GVHD through IDO-dependent modulation of inflammation.
Methods
Mice
B10.BR (H-2Kk), C57BL/6 (B6, H-2Kb), and FVB (H-2Kq) mice, as well as congenic strains of B6 expressing CD45.1 or CD90.1, and IFN-γ, IFN-γ receptor, and IDO1 knockout strains on the B6 background (IFN-γ−/−, IFNGR1−/−, and IDO1−/−), were purchased from The Jackson Laboratory. A congenic strain of B10.BR (H-2Kk) expressing CD90.1, named BA.B10, was generated by crossing B6 CD90.1 and B10.BR mice and then backcrossing 10 generations to the parental B10.BR strain at Emory University. Green fluorescent protein (GFP)–expressing B6 mice were a gift from Dr Robert Taylor (Emory University). Luciferase-expressing L2G85 mice on a FVB background were a gift from Dr Robert Negrin (Stanford University).12 Mice were used at 8 to 12 weeks of age. All procedures were carried out under a protocol approved by the Institutional Animal Care and Use Committee at Emory University.
Tumor cells
LBRM 33-5A4, a B10.BR T-cell lymphoma cell line,13 was purchased from ATCC, cultured according to ATCC recommendations, and tested to be free of lymphocytic choriomeningitis virus, mouse hepatitis virus, mouse minute virus, and mouse parvovirus by the University of Missouri Research Animal Diagnostic Laboratory. This cell line also was transfected to express luciferase in our laboratory, and the luciferase+ strain was used in an in vivo bioluminescent imaging experiment.
FACS sorting of HSC and pre-pDCs
Donor mice were killed in a humane manner, and femurs and tibias were removed aseptically. BM cells were harvested with sterile RPMI-1640 containing 1% heat-inactivated fetal calf serum (RPMI/FCS). Hematopoietic stem cells (HSCs) and pre-pDCs were sorted from bone marrow simultaneously using the following strategy. All antibodies were purchased from BD Biosciences Pharmingen unless otherwise noted. Bone marrow cells were stained with a cocktail of biotinylated antibodies (lineage 1 = CD3, CD11b, CD19, CD49b, IgM, and Ter119), followed by staining with streptavidin APC-Cy7 and a second lineage cocktail of PE-labeled antibodies (lineage 2 = CD4, CD8, GR-1, and I-Ab), as well as CD11c FITC, B220 PE-Cy5, C-kit APC, Sca-1 PE-Cy7, and PDCA1-Efluor450 (eBioscience). Cells were then sorted by FACS10 using an FACSAria cell sorter and Diva Version 5.1 software (both from BD Biosciences). After initial scatter-based gating to exclude doublets, the B220+ and B220− populations were identified and further gated for pre-pDCs and HSC sorting, respectively. Pre-pDCs were defined as B220+, lineage 1−, CD11c+, PDCA1+. HSCs were defined as B220−, lineage 1− (with less stringent gating than used for the pDC sort), lineage 2−, C-kit+ and Sca-1+ (Figure 1A). BM from 1 donor mouse typically provided sufficient sorted pre-pDCs and HSCs to transplant 3 recipients.

Purification, homing, and in vivo proliferation of donor pDCs after allogeneic transplantation. (A) Gating strategy for sorting both HSCs and pre-pDCs from mouse bone marrow. After initial scatter-based gating to exclude doublets, the B220+ and B220− populations were identified and further gated for pre-pDC and HSC sorting, respectively. Pre-pDCs were defined as B220+, lineage 1− (lineage 1 = CD3, CD11b, CD19, CD49b, IgM, and Ter119), CD11c+, and PDCA1+. Postsort analysis is shown in the top right plot. HSCs were defined as lineage 1− (with less stringent gating than used for the pDC sort), lineage 2− (lineage 2 = CD4, CD8, GR-1, and I-Ab), Sca-1+ and C-kit+. Postsort HSC analysis is shown in the third plot, bottom row. Analysis of CD4 and CD8 content of T cells purified by negative immunomagnetic selection (without prior gating) is shown in the bottom right plot. (B) Bioluminescent imaging images of pDCs trafficking in syngeneic and allogeneic recipients. FVB (H-2q; n = 5) and BA.B10 (CD90.1+, H-2k; n = 5) recipients were lethally irradiated and injected with pre-pDCs sorted from L2G85 luciferase+ mice in combination with FVB HSCs and T cells. Bioluminescent imaging images were taken weekly; representative images are shown. The abdomens of black-furred B10.BR allogeneic recipients (bottom panels) were depilidated with Nair before bioluminescent imaging.
Purification, homing, and in vivo proliferation of donor pDCs after allogeneic transplantation. (A) Gating strategy for sorting both HSCs and pre-pDCs from mouse bone marrow. After initial scatter-based gating to exclude doublets, the B220+ and B220− populations were identified and further gated for pre-pDC and HSC sorting, respectively. Pre-pDCs were defined as B220+, lineage 1− (lineage 1 = CD3, CD11b, CD19, CD49b, IgM, and Ter119), CD11c+, and PDCA1+. Postsort analysis is shown in the top right plot. HSCs were defined as lineage 1− (with less stringent gating than used for the pDC sort), lineage 2− (lineage 2 = CD4, CD8, GR-1, and I-Ab), Sca-1+ and C-kit+. Postsort HSC analysis is shown in the third plot, bottom row. Analysis of CD4 and CD8 content of T cells purified by negative immunomagnetic selection (without prior gating) is shown in the bottom right plot. (B) Bioluminescent imaging images of pDCs trafficking in syngeneic and allogeneic recipients. FVB (H-2q; n = 5) and BA.B10 (CD90.1+, H-2k; n = 5) recipients were lethally irradiated and injected with pre-pDCs sorted from L2G85 luciferase+ mice in combination with FVB HSCs and T cells. Bioluminescent imaging images were taken weekly; representative images are shown. The abdomens of black-furred B10.BR allogeneic recipients (bottom panels) were depilidated with Nair before bioluminescent imaging.
Donor T-cell purification
Donor mice were euthanized and spleens collected. Splenocytes were removed by perfusion of the spleen and gentle teasing with forceps. T cells were purified by incubating splenocytes with biotinylated anti-CD11b, B220, CD49b, and Ter119 antibodies, followed by anti-biotin microbeads (Miltenyi Biotec), and negative immunomagnetic selection (MACS) using an LS column (Miltenyi Biotec).
Transplantation
Recipient B10.BR or BA.B10 mice were irradiated with 2 doses of 5.5 Gy separated by 3 hours on day −2.14 On day 0, recipient mice were transplanted with combinations of 3 to 5 × 103 FACS-sorted HSCs, 5 × 104 FACS-sorted donor pre-pDCs, and 3 × 105 or 1 × 106 MACS-purified T cells from B6 CD45.1 donors. Mice were weighed twice weekly and examined daily for signs of GVHD as described previously.11 Moribund animals losing more than 25% of initial body weight, and mice surviving until the end of the experiment, were euthanized and tissues were processed for histopathologic analysis of tumor-trophic sites, including liver, small bowel, and large bowel. Flow cytometric chimerism analyses were performed on blood leukocytes on days 40 (± 1), 60 (± 2), and 90 (± 5) after transplant.
Posttransplant analysis of donor-derived T cells and pDCs
For simultaneous staining of Foxp3 and intracellular cytokine expression in donor-derived T cells, splenocytes were isolated from transplant recipients, incubated with phorbol 12-myristate 13-acetate/ionomycin and brefeldin A for 5 hours, and then labeled with antibodies against cell surface markers (CD45.1, CD3, CD4, CD8, and CD25), followed by intracellular staining methods using antibodies against cytokine targets (BD Biosciences Pharmingen) and anti–mouse Foxp3 (eBioscience) per the manufacturers' instructions. Simultaneous staining using antibodies against Foxp3, Granzyme, and Perforin was carried out in a similar manner without stimulation. Appropriate isotype controls were used for all intracellular targets. For detection of intracellular IDO in donor GFP+ pDCs, cells from recipient spleen or bone marrow were fixed and permeabilized using reagents from BD Biosciences Pharmingen, and intracellular expression of IDO in GFP+ pDCs was detected using anti-IDO monoclonal antibody clone 10.1 (Millipore Bioscience Research Reagents), followed with an appropriate labeled secondary antibody. For analysis of IDO expression in pDCs from cell culture experiments, cells were first stained with antibodies to identify pDCs (CD11c and B220), followed by fixation and permeabilization and staining with mIDO48 anti-IDO (BioLegend) using methods similar to those used by Mateoli et al with an appropriate fluorescently labeled secondary antibody.15
RT-PCR of IDO
Total RNA was extracted from FACS-purified pDCs by TRIzol reagent (Invitrogen), and RT was performed by SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) following the manufacturer's instructions. IDO PCR primers used were as described previously.16 The murine GAPDH housekeeping gene was used as an internal control. Real-time PCR was performed using Platinum SYBR Green quantitative PCR SuperMix-UDG (Invitrogen) on an ABI 7000 instrument (Applied Biosystems) using a primer-specific standard curve with denaturation (95°C for 10 minutes), amplification repeated 40 times (95°C for 30 seconds, 60°C for 40 seconds, 72°C for 30 seconds). For each sample, ddCt (crossing point) values were calculated as the Ct of the target gene minus the Ct of the GAPDH gene. Semiquantitative RT-PCR was performed (30 cycles; annealing temperature, 60°C) using primers described previously.16,17 PCR products were analyzed on a 1% agarose gel.
T-cell proliferation
The proliferation of donor T cells in recipient spleen was analyzed by CFSE dilution as described previously.18,19 In brief, donor T cells were stained with CFSE before transplant, and recipient spleens were removed 3 days later for preparation of cell suspensions. Proliferation of donor T cells was determined by flow cytometric analysis of CFSE dilution profiles gated on donor T-cell populations using FlowJo Version 9.1 software (TreeStar). The proliferation index (PI) for gated CD4 or CD8 T cells was determined using the CFSE histograms as described by Wallace and Muirhead as follows: the number of events (A) in each generation (k) are added together and divided by the total number of precursors, which is calculated for each generation by dividing the number of events in that generation by 2k.20
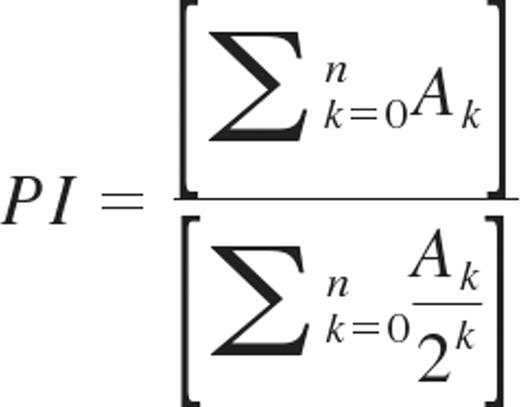
In vivo bioluminescent imaging
Bioluminescent imaging was performed as described previously.21,22 In brief, anesthetized mice were injected intraperitoneally with luciferin (10 μg/g body weight). Ten minutes later, mice were imaged using an IVIS100 charge-coupled device imaging system (Xenogen). Imaging data were analyzed with Living Image Version 3.2 software (Xenogen).
Statistical analyses
Analyses of data were performed using SPSS Version 17 for Mac. Data are presented as mean ± SEM. Survival differences between groups were calculated with the Kaplan-Meier log rank test in a pairwise manner. Differences in T-cell CFSE division numbers between groups and other parametric tests comparing multiple groups were performed using the 1-way ANOVA. Differences in repeated measurements of GVHD outcome between groups were determined using the Kruskal-Wallis nonparametric ANOVA test. Comparisons among numbers of T cells and cytokine positive T cells recovered in the spleens of transplant recipients were performed using the nonparametric Mann-Whitney U test. P values < .05 were considered significant.
Results
Donor pDCs home to, persist, and expand in the lymph nodes and spleen of transplant recipients
A FACS gating strategy was developed to concurrently sort pre-pDCs and HSCs from a mouse bone marrow suspension (Figure 1A). To define the kinetics of donor pDC persistence and expansion after transplant, we sorted pre-pDCs from luciferase+ L2G85 transgenic mice (FVB background) and transplanted 1 × 105 luciferase+ pre-pDCs in combination with 3 × 103 C-kit+ Sca-1+ HSCs and 3 × 105 T cells from FVB nontransgenic donors into FVB (syngeneic) or B10.BR (allogeneic) mice after lethal irradiation. Of note, HSCs were specifically excluded from the pre-pDCs sorted population (B220+, lineage 1− CD11c+ PDCA1+) because HSCs do not express PDCA-1. Bioluminescent imaging showed that luciferase+ donor pDCs were detected at 2 weeks after transplantation and that the numbers of luciferase+ donor pDCs expanded in the lymph nodes and spleen of transplant recipients over the next week but that they did not contribute to long-term donor hematopoietic cell engraftment (Figure 1B).
IFN-γ synthesis by donor T cells limits GVHD
IFN-γ has been shown to be involved both in alloreactive GVL effects as well as in limiting GVHD severity.23,24 Using allogeneic MHC-mismatched HSCT (C57BL/6→B10.BR) in mice harboring the T lymphoblastic leukemia cell line LBRM, we have reported previously that recipients transplanted with FACS-purified lineage− CD11c+ CD11b− DCs in combination with purified HSCs and T cells had better leukemia-free survival, higher numbers of IFN-γ–producing donor T-cells, as well as higher levels of serum IFN-γ than mice transplanted with HSCs and T cells alone.10 Here, we tested whether production of IFN-γ by donor T cells was necessary for the improved leukemia-free survival using lineage− CD11c+ CD11b− PDCA1+ B220+ donor pre-pDCs.10 Similar to the results of Yang et al,25 transplantation of T cells from allogeneic IFN-γ knockout (IFN-γ−/−) mice led to severe acute GVHD compared with recipients of T cells from wild-type mice. B10.BR recipients that received 3 × 103 C57BL/6 wild-type HSCs and 3 × 105 T cells from IFN-γ knockout donors died rapidly because of GVHD, with 0% survival at day 60 compared with 70% survival among recipients of the same numbers of wild-type donor T cells (Figure 2A; P < .01). Addition of 5 × 104 donor pre-pDCs to these grafts resulted in survival rates similar to the corresponding groups without pre-pDCs (Figure 2A). In tumor-bearing transplant recipients, infusion of wild-type donor T cells and pre-pDCs resulted in 65% survival compared with 0% survival for recipients of IFN-γ−/− T cells with or without added pre-pDCs (Figure 2B; P < .01), supporting a role for donor pDCs in enhancing the antitumor effect of donor T cells. The addition of wild-type donor pre-pDCs to grafts containing wild-type T cells led to a transient reduction in GVHD scores compared with recipients of wild-type T cells and IFN-γ−/− donor T cells only in nontumor-bearing mice (Figure 2C). It is also noteworthy that donor pre-pDCs did not augment GVHD in either the tumor or nontumor transplant models (Figure 2C-D).

IFN-γ–deficient allogeneic donor T cells induce severe GVHD without enhanced GVL effects. Survival (A-B) and mean GVHD clinical scores (C-D) of mice that received 3 × 103 FACS-sorted HSCs + 3 × 105 MACS-enriched splenic T cells from with either WT or IFN-γ knockout (IFN-γ−/−) mice with or without 5 × 104 FACS-purified pDCs in C57BL/6J→CD90.1+ BA.B10 transplant model. Leukemic recipients also were injected with 1 × 105 viable LBRM tumor cells on day −1, 1 day after radiation and 1 day before BMT. n = 10 mice per group in panel A and n = 20 mice per group in panel B. **P < .01 comparing HSCs + WT T with HSCs + WT T + pDCs for days 13-22; #P < .05 comparing HSCs + WT T with HSCs + WT T + pDCs for days 44-58; *P < .05 comparing HSCs + WT T with HSCs + IFN-γ−/− T for days 22-33 in panel C; and ***P = < .001 comparing HSCs + WT T with HSCs + IFN-γ−/− T for day 19 after transplantation; *P = < .05 comparing HSCs + WT T with HSCs + IFN-γ−/− T for days 22-41 after transplantation; $P = < .05 comparing HSCs + WT T with HSCs + WT T + pDCs for days 26-54 after transplantation; #P < .01 comparing HSCs + WT T with HSCs + IFN-γ−/− T + pDCs for days 13-26 in panel D.
IFN-γ–deficient allogeneic donor T cells induce severe GVHD without enhanced GVL effects. Survival (A-B) and mean GVHD clinical scores (C-D) of mice that received 3 × 103 FACS-sorted HSCs + 3 × 105 MACS-enriched splenic T cells from with either WT or IFN-γ knockout (IFN-γ−/−) mice with or without 5 × 104 FACS-purified pDCs in C57BL/6J→CD90.1+ BA.B10 transplant model. Leukemic recipients also were injected with 1 × 105 viable LBRM tumor cells on day −1, 1 day after radiation and 1 day before BMT. n = 10 mice per group in panel A and n = 20 mice per group in panel B. **P < .01 comparing HSCs + WT T with HSCs + WT T + pDCs for days 13-22; #P < .05 comparing HSCs + WT T with HSCs + WT T + pDCs for days 44-58; *P < .05 comparing HSCs + WT T with HSCs + IFN-γ−/− T for days 22-33 in panel C; and ***P = < .001 comparing HSCs + WT T with HSCs + IFN-γ−/− T for day 19 after transplantation; *P = < .05 comparing HSCs + WT T with HSCs + IFN-γ−/− T for days 22-41 after transplantation; $P = < .05 comparing HSCs + WT T with HSCs + WT T + pDCs for days 26-54 after transplantation; #P < .01 comparing HSCs + WT T with HSCs + IFN-γ−/− T + pDCs for days 13-26 in panel D.
IFN-γ synthesis by donor T cells limits the activation and proliferation of donor CD8+ T cells
We next used CFSE-labeled T cells to test the effects of IFN-γ synthesis on early alloreactive donor T-cell proliferation. Lethally irradiated BA.B10 mice received 3 × 105 CFSE-labeled T cells from either wild-type or IFN-γ−/− B6 donors, in combination with FACS-sorted 3 × 103 HSCs and 5 × 104 pre-pDCs. Recipients were euthanized at day 3, splenocytes were prepared, and donor T-cell proliferation was assessed by measuring CFSE dilution using flow cytometry (representative CFSE profiles from each group are shown in Figure 3A). Wild-type donor CD8+ T cells underwent more initial cell divisions than CD4+ T cells, and IFN-γ−/− T cells underwent more cell divisions compared with wild-type T cells (Figure 3A-B). The addition of donor pDCs to the combination of HSCs and wild-type T cells enhanced the initial proliferation of donor T cells, and decreased the fraction of nondivided donor T cells, reflected in an increased proliferation index (P < .05; Figure 3B-C).

Absence of IFN-γ synthesis by donor T cells leads to enhanced cell division among donor CD8+ T cells. CD90.1+ BA.B10 mice received 3 × 103 FACS-sorted HSCs and 3 × 105 CFSE-labeled CD90.2+ T cells from WT (closed symbols) or IFN-γ−/− (open symbols) B6 donors with or without 5 × 104 FACS-purified pDCs (n = 3/group). Recipient splenocytes were prepared at day 3 after transplant and analyzed for the proliferation of donor T cells. (A) Representative CFSE proliferation profiles of CD4 and CD8 T cells for each transplant combination. Gating indicates the average percentage of cells in the undivided T-cell peak. (B) Average percentage of cells in each division peak (CD4 panel, ***P < .001 comparing percentage of undivided T cells in HSCs + WT T vs all other groups; *P < .05 comparing percentage of T cells divided 5 times for HSCs + WT T vs HSCs + IFN-γ−/− T + pDCs and P < .05 comparing percentage of T cells divided 6 times for HSCs + WT T vs HSCs + WT T + pDCs; CD8 panel, ***P < .001 comparing percentage of undivided T cells and T cells divided 1 or 2 times in HSCs + WT T vs all other groups; *P < .05 comparing percentage of T cells divided 5 or 6 times for HSCs + WT T vs HSC + IFN-γ−/− T and divided 6 or 7 times for HSCs + WT T vs HSCs + IFN-γ−/− T + pDCs). (C) Calculated proliferation indices (*P ≤ .05 compared with HSCs + T group by 1-tailed Mann-Whitney U test).
Absence of IFN-γ synthesis by donor T cells leads to enhanced cell division among donor CD8+ T cells. CD90.1+ BA.B10 mice received 3 × 103 FACS-sorted HSCs and 3 × 105 CFSE-labeled CD90.2+ T cells from WT (closed symbols) or IFN-γ−/− (open symbols) B6 donors with or without 5 × 104 FACS-purified pDCs (n = 3/group). Recipient splenocytes were prepared at day 3 after transplant and analyzed for the proliferation of donor T cells. (A) Representative CFSE proliferation profiles of CD4 and CD8 T cells for each transplant combination. Gating indicates the average percentage of cells in the undivided T-cell peak. (B) Average percentage of cells in each division peak (CD4 panel, ***P < .001 comparing percentage of undivided T cells in HSCs + WT T vs all other groups; *P < .05 comparing percentage of T cells divided 5 times for HSCs + WT T vs HSCs + IFN-γ−/− T + pDCs and P < .05 comparing percentage of T cells divided 6 times for HSCs + WT T vs HSCs + WT T + pDCs; CD8 panel, ***P < .001 comparing percentage of undivided T cells and T cells divided 1 or 2 times in HSCs + WT T vs all other groups; *P < .05 comparing percentage of T cells divided 5 or 6 times for HSCs + WT T vs HSC + IFN-γ−/− T and divided 6 or 7 times for HSCs + WT T vs HSCs + IFN-γ−/− T + pDCs). (C) Calculated proliferation indices (*P ≤ .05 compared with HSCs + T group by 1-tailed Mann-Whitney U test).
IDO is up-regulated in pDCs responding to alloantigen
One of the IFN-γ–inducible genes, IDO, has been linked to the regulation of immune responses after transplantation.26,27 We sought to determine whether IDO expression is up-regulated in donor pDCs in our allogeneic transplant model. We first tested the effect of exogenous IFN-γ in cultured pDCs. As measured by intracellular staining (Figure 4A), semiquantitative RT-PCR(Figure 4B), and real-time quantitative RT-PCR (data not shown),28,29 IFN-γ induced IDO expression in pDCs freshly isolated from donor bone marrow. We then measured the expression of IDO in donor pDCs in an allogeneic transplant setting in which GFP+ pre-pDCs were administered with wild-type donor HSCs and T cells, and bone marrow cells and splenocytes from transplant recipients were recovered for flow cytometric analysis 10 days after transplant. IDO was up-regulated in GFP+ pDCs that were recovered from spleens of allogeneic transplant recipients but not in GFP+ pDCs recovered from bone marrow (Figure 4C), suggesting that IDO up-regulation in donor pDCs required synthesis of IFN-γ by donor T cells responding to alloantigen in a lymphoid organ. This conclusion was supported by results of in vitro experiments demonstrating that the presence of both T cells and alloantigen was required to up-regulate IDO in pDCs cultured for 3 days, with 27% of pDCs in cultures containing wild-type T cells and allogeneic irradiated splenocytes expressing IDO (Figure 4D; specific staining-isotype staining). IDO up-regulation in pDCs did not occur without the addition of alloantigen, or in donor pDCs that lack the IFN-γ receptor (IFNGR1−/−) or negative control pDC from IDO1−/− mice (Figure 4D).

IFN-γ and immune response to alloantigen up-regulate IDO in pDCs. (A) Pre-pDCs from WT B6 BM were exposed to 100 ng/mL IFN-γ for 18 hours, and IDO expression was measured by FACS (heavy line). Pre-DCs from WT mice without IFN-γ treatment and pre-pDCs from IDO1−/− mice were used as controls. Mean fluorescence intensity (MFI) of intracellular IDO staining for each condition is shown in the table. (B) mRNA levels of pDCs were also assessed by semiquantitative RT-PCR with GAPDH as an internal control. (C) B6→BA.B10 transplant recipients of HSCs, T cells, and 5 × 105 GFP+ pre-pDCs were killed at day 10, bone marrow and spleen cells were harvested, and IDO expression in donor GFP+ pDCs was detected by intracellular staining (n = 3). (D) FACS-purified pre-pDCs from WT, IFNGR1−/−, or IDO1−/− bone marrow were cultured with WT T cells with or without irradiated B10.BR splenocytes (Ag) in 96-well round-bottomed plates, and IDO expression was detected by intracellular staining on day 3. MFI for IDO staining is shown in the table. Experiments for panels A and D were performed twice, with data shown from 1 experiment.
IFN-γ and immune response to alloantigen up-regulate IDO in pDCs. (A) Pre-pDCs from WT B6 BM were exposed to 100 ng/mL IFN-γ for 18 hours, and IDO expression was measured by FACS (heavy line). Pre-DCs from WT mice without IFN-γ treatment and pre-pDCs from IDO1−/− mice were used as controls. Mean fluorescence intensity (MFI) of intracellular IDO staining for each condition is shown in the table. (B) mRNA levels of pDCs were also assessed by semiquantitative RT-PCR with GAPDH as an internal control. (C) B6→BA.B10 transplant recipients of HSCs, T cells, and 5 × 105 GFP+ pre-pDCs were killed at day 10, bone marrow and spleen cells were harvested, and IDO expression in donor GFP+ pDCs was detected by intracellular staining (n = 3). (D) FACS-purified pre-pDCs from WT, IFNGR1−/−, or IDO1−/− bone marrow were cultured with WT T cells with or without irradiated B10.BR splenocytes (Ag) in 96-well round-bottomed plates, and IDO expression was detected by intracellular staining on day 3. MFI for IDO staining is shown in the table. Experiments for panels A and D were performed twice, with data shown from 1 experiment.
IDO expression in donor pDCs limits the GVHD activity of donor T cells without inhibiting the irGVL activity
To determine whether the ability of donor pDCs to limit GVHD might be attributable to the expression of IDO, we used IDO knockout (IDO1−/−) mice as donors of FACS-purified pDCs in the C57BL/6→B10.BR transplant model. As shown in Figure 5A, survival after a nonlethal dose of donor T cells (3 × 105) was significantly worse in the presence of IDO1−/− donor pDCs compared with wild-type pDC (P = .003), and the addition of donor IDO1−/− pDCs increased the severity of GVHD, as depicted by GVHD scores (Figure 5B; P < .01 comparing recipients of IDO1−/− vs wild-type pDCs, and P < .05 comparing IDO1−/− pDCs + T cells to recipients of T cells alone). At day 10 after transplant, considerable expansion of donor graft-derived T cells was evident by the numbers of circulating donor T cells in all 3 groups (Figure 5C), with the highest numbers in recipients of wild-type pDCs, fewer in recipients of IDO1−/− pDCs, and lowest numbers in recipients of HSCs + T cells alone (P = NS because of variation between samples). The expansion of mature donor graft T cells was transient, with much lower numbers seen at day 40, concomitant with a shift to HSC-derived thymopoiesis and the appearance of HSC-derived donor T cells (Figure 5D). The levels of donor graft-derived T cells at later time points were lower than what we have reported in previous studies,10 perhaps because of the rigorous purification of donor pre-pDCs performed here, and the elimination of the donor HSCs that contaminated the CD11b− APC population that may have contributed to donor T-cell chimerism described in our previous work. Recipient-type T cells were present at minimal numbers in the blood over the course of the experiment (Figure 5E). Histologic analyses of target organs of GVHD from mice killed on day 10 showed that most mice in all the treatment groups had detectable GVHD in the liver or intestine, with the highest average GVHD histology score seen in the group that received HSCs + T cells and IDO1−/− pre-pDCs, but intergroup differences in the histologic GVHD score were not significant because of small numbers of animals evaluated at this time point (data not shown).

IDO-deficient donor pDCs induce more severe GVHD activity. Survival (A) and mean GVHD scores (B) of mice that received 3 × 103 HSCs + 3 × 105 B6 splenic T cells (n = 15), and groups that also received 5 × 104 pre-pDCs from WT donors (n = 15) or IDO1−/− donors (n = 12), in the C57BL/6J→BA.B10 transplant model. Data are means ± SEM from 3 replicate experiments with 5 to 6 mice per group. (C-E) T-cell chimerism analyses of animals from panel A, showing blood T cells derived from mature T cells in the graft (C), donor HSC-derived T cells (D), and residual host T cells (E). Day 0 values for host T cells were determined using 3 mice that were irradiated and then bled 2 days later, with absolute T-cell levels averaging 3% of control. Day 10 values were determined from a separate experiment with recipient mice killed at this time point (n = 3). Survival (F) of mice transplanted using the same conditions as in panel A and also injected with 1 × 105 viable LBRM tumor cells at day −1. Data from 4 independent experiments are combined. n = 30 mice in both the HSCs + T cell and the HSCs + T + WT pre-pDC groups and n = 9 in the HSC + T + IDO1−/− pre-pDC group (*P < .01 comparing recipients of T cells and WT pre-pDCs with recipients of T cells and IDO1−/− pre-pDCs).
IDO-deficient donor pDCs induce more severe GVHD activity. Survival (A) and mean GVHD scores (B) of mice that received 3 × 103 HSCs + 3 × 105 B6 splenic T cells (n = 15), and groups that also received 5 × 104 pre-pDCs from WT donors (n = 15) or IDO1−/− donors (n = 12), in the C57BL/6J→BA.B10 transplant model. Data are means ± SEM from 3 replicate experiments with 5 to 6 mice per group. (C-E) T-cell chimerism analyses of animals from panel A, showing blood T cells derived from mature T cells in the graft (C), donor HSC-derived T cells (D), and residual host T cells (E). Day 0 values for host T cells were determined using 3 mice that were irradiated and then bled 2 days later, with absolute T-cell levels averaging 3% of control. Day 10 values were determined from a separate experiment with recipient mice killed at this time point (n = 3). Survival (F) of mice transplanted using the same conditions as in panel A and also injected with 1 × 105 viable LBRM tumor cells at day −1. Data from 4 independent experiments are combined. n = 30 mice in both the HSCs + T cell and the HSCs + T + WT pre-pDC groups and n = 9 in the HSC + T + IDO1−/− pre-pDC group (*P < .01 comparing recipients of T cells and WT pre-pDCs with recipients of T cells and IDO1−/− pre-pDCs).
Using the same numbers of donor cells transplanted into LBRM tumor-bearing mice, 60% of mice transplanted with wild-type pre-pDCs were long-term survivors, whereas only 15% of the recipients of HSCs + T cells survived to day 80 (Figures 5F; P = .002 comparing recipients of IDO1−/− pDC vs wild-type pre-pDCs), and all recipients of pre-pDCs from IDO1−/− mice died by day 60. Thus, the presence of wild-type donor pre-pDCs in the graft augmented the GVL activity of donor T cells without increasing their GVHD activity. Using mice bearing luciferase-transfected LBRM and bioluminescent imaging as well as histologic examination of samples of liver and intestine obtained at necropsy of mice from experiments that included LBRM, we found gross evidence of tumor cell infiltration in 23% of recipients of HSCs + T cells, and none of the mice that received the combination of wild-type pre-pDCs, HSCs, and T cells, or IDO1−/− pre-pDCs, HSCs, and T cells (data not shown).
We next tested the effect of adding donor pDCs from wild-type and IDO1−/− mice to a dose of donor T cells (1 × 106) that we have previously shown to cause significant GVHD-related posttransplant mortality in B6→B10.BR transplants.11 The experimental group that received the combination of HSCs + 1 × 106 T cells had 14% long-term survival, with deaths attributed to GVHD (Figure 6A-B). Remarkably, the addition of wild-type donor pre-pDCs protected mice from lethal GVHD, with 51% day 80 survival. Notably, the protective effect of donor pre-pDCs was abrogated when pre-pDCs were purified from IDO1−/− donors, as shown by decreased survival (Figure 6A; P = .002) and increased GVHD scores (Figure 6B; P < .0006) compared with recipients of wild-type pre-pDCs. Addition of pre-pDCs from IDO1−/− donors led to enhanced GVHD mortality, with a median survival of 12 days and no survival beyond day 43 after transplant. These data indicate a major role for IDO expression by donor pre-pDCs in limiting GVHD in this model system.

WT pDC limit the GVHD activity of donor T cells while enhancing donor T-cell proliferation. (A) Survival of mice that received 3 × 103 FACS-sorted HSCs + 1 × 106 MACS-enriched B6 splenic T cells with or without 5 × 104 FACS-purified pDCs from either WT or IDO1−/− mice in the B6→BA.B10 transplant model (*P = .002 comparing WT pDC and IDO1−/− pDC recipient groups). (B) GVHD scores for these groups (**P < .0006 comparing WT pDC and IDO1−/− pDC recipient groups). Data are means ± SEM from 4 replicate experiments with average 6 mice per group. (C) B10.BR mice received 5 × 103 FACS-sorted B6 HSCs and 1 × 106 B6 (CD45.1+) T cells, with or without addition of 5 × 104 FACS-purified pre-pDCs from WT or IDO1−/− B6 donors. The experiment was performed 3 times with 2 to 3 mice per group, for a total n = 7 for the HSC + T group and n = 8 for the groups with added WT pDCs or IDO1−/− pDCs. T cells were CFSE labeled before transplant. Proliferation profiles for donor CD45.1+ CD4 and CD8 T cells recovered from recipient spleens on day 3.5 after transplant are shown for representative samples, with gates indicating the percentages of cells that remained undivided (right) or that were in divisions 1 to 6 (center) or later divisions (left). Bar graphs show the average percentage of undivided CD4 or CD8 T cells in the CFSE profile and the calculated proliferation indices. (D) Representative flow cytometry plots gated on donor CD4 T cells from recipient spleens on day 3.5 after transplant, with boxes indicating the CD25+ FoxP3+ T-reg population. (E) Comparisons of the absolute numbers of donor graft-derived CD8 and CD4 T cells and CD4+CD25+FoxP3+ T-regs present in recipient spleens on day 3.5 after transplant.
WT pDC limit the GVHD activity of donor T cells while enhancing donor T-cell proliferation. (A) Survival of mice that received 3 × 103 FACS-sorted HSCs + 1 × 106 MACS-enriched B6 splenic T cells with or without 5 × 104 FACS-purified pDCs from either WT or IDO1−/− mice in the B6→BA.B10 transplant model (*P = .002 comparing WT pDC and IDO1−/− pDC recipient groups). (B) GVHD scores for these groups (**P < .0006 comparing WT pDC and IDO1−/− pDC recipient groups). Data are means ± SEM from 4 replicate experiments with average 6 mice per group. (C) B10.BR mice received 5 × 103 FACS-sorted B6 HSCs and 1 × 106 B6 (CD45.1+) T cells, with or without addition of 5 × 104 FACS-purified pre-pDCs from WT or IDO1−/− B6 donors. The experiment was performed 3 times with 2 to 3 mice per group, for a total n = 7 for the HSC + T group and n = 8 for the groups with added WT pDCs or IDO1−/− pDCs. T cells were CFSE labeled before transplant. Proliferation profiles for donor CD45.1+ CD4 and CD8 T cells recovered from recipient spleens on day 3.5 after transplant are shown for representative samples, with gates indicating the percentages of cells that remained undivided (right) or that were in divisions 1 to 6 (center) or later divisions (left). Bar graphs show the average percentage of undivided CD4 or CD8 T cells in the CFSE profile and the calculated proliferation indices. (D) Representative flow cytometry plots gated on donor CD4 T cells from recipient spleens on day 3.5 after transplant, with boxes indicating the CD25+ FoxP3+ T-reg population. (E) Comparisons of the absolute numbers of donor graft-derived CD8 and CD4 T cells and CD4+CD25+FoxP3+ T-regs present in recipient spleens on day 3.5 after transplant.
Donor pre-pDCs enhanced initial donor T-cell proliferation of donor T cells
To explore the mechanism by which wild-type donor pDCs might modulate the GVHD activity of a lethal dose of donor T cells, we tested their effect on early T-cell proliferation 3.5 days after transplant, and we contrasted the results seen with cotransplanting T cells with IDO−/− pDCs or no additional donor pDCs. B10.BR mice were lethally irradiated and transplanted with 5 × 103 C-kit+ Sca-1+ HSCs and a high dose of CFSE-labeled donor T cells (1 × 106) with or without addition of 5 × 104 FACS-purified pre-pDCs from wild-type C57BL/6 or IDO1−/− donor mice. Proliferation of donor T cells recovered from the spleens of transplant recipients was determined on posttransplant day 3.5. Similar to the results using a lower dose of donor T cells shown in Figure 3, analysis of CFSE profiles (Figure 6C) and determination of absolute numbers of donor CD4 and CD8 T cells in recipient spleens showed greater T-cell proliferation in the presence of wild-type donor pre-pDCs compared with other transplant groups (Figure 6E), although the differences were only marginally significant (eg, P = .1 comparing percentages of undivided CD4 T cells in the CFSE profiles for recipients of wild-type pre-pDCs vs IDO1−/− pre-pDC. Analysis of Foxp3 expression in CD4+CD25+ donor T cells also showed a trend toward larger numbers of donor T-reg among recipients of wild-type pDCs (Figure 6D-E; P = NS), and examination of the CFSE profiles of these T-regs showed CFSE fluorescence intensities corresponding to 7 or more cell divisions (data not shown). The average frequency of donor T-regs on day 3.5 after transplant among recipients of wild-type pre-pDCs (11.4% of donor CD4+ T cells) was similar to the frequency of T-regs before transplantation (10.5% of CD4+ T cells).
IDO expression in donor pre-pDCs altered the balance between donor T-reg and inflammatory donor T cells
To further explore the mechanism by which donor pre-pDCs modulate the GVHD activity of donor T cells and why pDCs from IDO1−/− donor mice failed to limit GVHD, we used the same model system and studied the immune polarization of donor T cells on day 10 after transplant. To avoid early loss of mice from hyperacute GVHD in these experiments, we used a lower dose of T cells (3 × 105) that we have shown not to cause early GVHD-related death (Figure 3). We used a panel of antibodies to interrogate intracellular expression of cytokines, perforin, granzyme, and foxp3 to determine the immune polarization, cytolytic activity, and foxp3 expression of CD4+ and CD8+ T cells of donor graft T cells in splenocytes recovered on day 10 after transplant from recipient mice. The total numbers of donor CD4+ and CD8+ T cells recovered from the spleens of transplant recipients were similar comparing recipients of HSCs + T cells with recipients of HSCs, T cells, and either wild-type or IDO1−/− pre-pDCs (Figure 7A). Of note, the average number of donor T cells in the spleens of the recipients of HSCs and T-cell grafts had increased ∼ 50- to 250-fold, from 29 000 donor CD4+ and 49 000 donor CD8+ T cells/spleen to 1.8 × 106 donor CD4+ and 13 × 106 donor CD8+ T cells/spleen over the ensuing 7 days. Similar degrees of donor T-cell expansion were seen in recipients of either wild-type or IDO1−/− pre-pDCs. Recipients of IDO1−/− pre-pDCs had less than half the numbers of foxp3+ CD4+ donor T-reg and perforin+ CD8+ T cells in their spleens compared with recipients of HSCs + T cells, although relatively large intraexperiment variance made the differences only marginally significant (Figure 7B; P = .1). The percentages of CD4+ and CD8+ donor T cells expressing IFN-γ or TNF-α were similar comparing the 3 treatment groups (Figure 7C-D) as were levels of expression for IL-2 and IL-10 (data not shown). IL-17 expression tended to be higher among CD4+ donor T cells from recipients of IDO1−/− pre-pDCs compared with recipients of HSCs + T cells (Figure 7E; P = NS), such that the ratio of IL17+ CD4+ T cells to foxp3+ CD4+ donor T-reg from recipients of IDO−/− pre-pDCs was 2-fold higher than for recipients of HSCs + T cells (Figure 7F).

Transplantation of IDO1−/− donor pDCs resulted in altered proportions of donor T-reg and inflammatory donor T cells. B10.BR mice were lethally irradiated and transplanted with 3 to 5 × 103 FACS-sorted HSCs (CD45.2+) and 3 × 105 T cells from B6 (CD45.1+) donors with 5 × 104 FACS-purified pDCs either from WT or IDO1−/− B6 donors. The experiment was performed 4 times with 3 to 5 recipients per group, for a total n = 14 for the HSCs + T group, n = 13 for the group with added WT pDCs, and n = 16 for recipients of IDO1−/− pDCs. Recipient spleens were harvested on day 10 after transplant for quantification of donor T-cell populations and cytokine expression analysis. (A-E) Averages and SEM for each group. (A) Total numbers of CD4 and CD8 donor T cells in recipient spleens. (B) Total numbers of CD8 donor T cells expressing Granzyme and Perforin, and total numbers of donor CD4+CD25+FoxP3+ T-regs (Tregs). (C-E) Percentage of donor CD4 and CD8 T cells expressing IFN-γ (C), TNF-α (D), and IL-17 (E). (F) Ratio comparing the average numbers of IL-17+ CD4 T cells in recipient spleens to the average numbers of CD4+CD25+FoxP3+ T-regs.
Transplantation of IDO1−/− donor pDCs resulted in altered proportions of donor T-reg and inflammatory donor T cells. B10.BR mice were lethally irradiated and transplanted with 3 to 5 × 103 FACS-sorted HSCs (CD45.2+) and 3 × 105 T cells from B6 (CD45.1+) donors with 5 × 104 FACS-purified pDCs either from WT or IDO1−/− B6 donors. The experiment was performed 4 times with 3 to 5 recipients per group, for a total n = 14 for the HSCs + T group, n = 13 for the group with added WT pDCs, and n = 16 for recipients of IDO1−/− pDCs. Recipient spleens were harvested on day 10 after transplant for quantification of donor T-cell populations and cytokine expression analysis. (A-E) Averages and SEM for each group. (A) Total numbers of CD4 and CD8 donor T cells in recipient spleens. (B) Total numbers of CD8 donor T cells expressing Granzyme and Perforin, and total numbers of donor CD4+CD25+FoxP3+ T-regs (Tregs). (C-E) Percentage of donor CD4 and CD8 T cells expressing IFN-γ (C), TNF-α (D), and IL-17 (E). (F) Ratio comparing the average numbers of IL-17+ CD4 T cells in recipient spleens to the average numbers of CD4+CD25+FoxP3+ T-regs.
Discussion
The current study was designed to explore the mechanism by which donor pDCs communicate with donor T cells to regulate their GVHD and GVL activities. We demonstrate, for the first time, that bidirectional signaling between donor T cells and donor pDCs regulates donor T-cell activation, proliferation, and immune polarization. Using bioluminescent imaging, we have shown here that transplanted donor pDCs persisted and expanded in vivo over the first 3 weeks after transplant. In a previous study, we showed that transplanted donor pre-pDCs matured in vivo, up-regulated MHC II, and were in proximity to transplanted donor T cells. The addition of donor pre-pDCs to grafts composed of purified HSCs and T cells augmented the initial proliferation of donor T cells, and their production of IFN-γ.10 Here, we confirmed the effect of donor pre-pDCs on enhancing donor T-cell proliferation in vivo, and we found that IFN-γ synthesis by T cells limited donor T-cell proliferation in vivo and induced IDO synthesis in pDCs in vitro. We observed a striking increase in the severity of GVHD among recipients of either IFN-γ−/− donor T cells or IDO1−/− pre-pDCs. These data indicate that IFN-γ−/− and IDO-mediated cross-talk between donor T cells and donor pDCs limits the GVHD activity of donor T cells. For these experiments, we used rigorously purified populations of donor cells and transplanted defined numbers of congenically marked donor T cells, HSCs, and pDCs to isolate the contributions of each component of the graft to posttransplant immune reconstitution, including GVHD and GVL. Although this reductionist approach allows mechanistic insights into the early posttransplant events that occur in allogeneic HSCT, our results do not formally exclude the possibility that donor T cell–derived IFN-γ acts on cellular targets other than donor DCs to ameliorate GVHD, the contribution of host antigen-presenting cells to the regulation of GVHD in allogeneic BMT, or the effect of IFN-γ or IDO on the homing and activation status of donor T cells at the sites of GVHD in the skin, gut, or liver.30 These caveats notwithstanding, our results suggest a complex and dynamic relationship between donor T cells and donor pDCs that augments GVL and attenuates GVHD through the sequential induction of T-helper 1 (Th1) immunity in the hematolymphoid microenvironment, followed by activation of regulatory immune mechanisms mediated by donor DCs. These data also suggest that the immunologic effects of administering Th1 cytokines to transplant recipients may result in significantly disparate effects depending on the timing of administration. Th1 cytokines (IL-12 and IFN-γ) given before engraftment and differentiation of donor HSCs may affect the immunologic cross-talk between donor DCs and donor T cells, whereas the effect of later cytokine administration may depend more on the immunologic activities of HSC derivatives.
Although a paradoxical role of IFN-γ in both promoting and limiting GVHD has been described previously,24,31,32 the mechanism for the disparate effects of IFN-γ in allogeneic transplantation has not been clearly elucidated heretofore.33 IFN-γ has been reported to enhance the GVL activity of donor T cells and is also involved in limiting GVHD activities by increasing apoptosis of CD4+ and CD8+ T cells, regulating target gene expression, modulating the susceptibility of GVHD target tissues to damage by alloreactive T cells,32-35 and inducing IDO gene expression.26,27,36 Our data support a critical role for IFN-γ synthesis by donor T cells and IDO up-regulation by donor pDCs in limiting donor T-cell expansion and GVHD.29,37 In the current study, the absence of IDO expression by donor pDCs was associated with increased GVHD. Thus, IDO expression by donor pDCs is a critical downstream event of IFN-γ expression by donor T cells that can inhibit continued T-cell activation and GVHD.
An important aspect of the current study is that we analyzed donor T-cell immune function at very early times after transplant (3 and 10 days after transplant) to better appreciate the early posttransplant interactions between donor T cells and donor pDCs and the effects of transplanting IFN-γ−/− and IDO1−/− donor immune cells. Although IFN-γ−/− T cells clearly proliferated at higher rates than wild-type T cells, the effects of transplanting IDO1−/− pDCs on donor T-cell activation and proliferation were more subtle. We noted significant variations in the proportion of donor T cells expressing IL-17, IFN-γ, and TNF-α in experiments comparing recipients of wild-type and IDO1−/− pDCs. A technical limitation of these experiments was the large variations in the repopulation of the spleen by donor immune cells and hematopoietic progenitors, due, in part, to the stochastic nature of CFU-S homing to the spleen (evident in the current studies as grossly visible spleen colonies on day 10 after transplant).38 Acknowledging that significant variance in absolute numbers of donor cells limits the statistical power of comparisons between treatment groups, our data suggest that donor pDCs have an effect on expansion of donor T-reg expansion and homing at the day +3 time point, an effect that was less evident at the later time point. By day 10 after transplant, there was very significant contribution of donor HSC-derived hematopoiesis to repopulation of the spleen in transplant recipients, and the immunologic effect of the relatively small numbers of donor pre-pDCs in the graft on the proliferation and activation status of donor T cells may be diluted (or negated) by the immunologic effects of HSC-derived DCs. We did observe an overall increase in the numbers of donor T-reg in the spleens of transplanted mice between day 3 and day 10 after transplant in all treatment groups. To recover sufficient donor T cells for analysis at day 3 after transplant, we used grafts containing 1 000 000 CFSE-labeled mature T cells including ∼ 60 000 T-reg, and recovered up to 7000 T-reg (∼ 10% of the input number) from the recipient spleen. For the day 10 experiments, we used grafts of 300 000 CD45.1+ mature T cells containing ∼ 20 000 T-regs, and recovered 23 000 to 46 000 CD45.1+ donor T-regs in the recipient spleen on day 10, absolute numbers equal to or greater than the total number of T-regs transplanted in the graft. Although survival and redistribution of transplanted T-regs could potentially account for some of the increased numbers of T-regs seen in the spleen at day 10, the relative fraction of total donor T-regs transplanted to the numbers recovered in the spleen at the 2 time points indicates that some level of in vivo expansion has probably occurred. It is noteworthy that in transplant recipients of IDO1−/− pre-pDCs, donor CD4+ T cells showed a trend toward higher levels of IL-17 expression and lower numbers of T-regs on day 10 after transplant, suggesting that the IDO expression in donor pre-pDCs and pDCs may regulate the generation of polyfunctional inflammatory T cells, including Th17 cells and Tr1-like T cells.32,39,40 Significantly, although survival of tumor-bearing mice transplanted with wild-type donor T cells and pre-pDCs was enhanced, consistent with augmentation of the GVL activity of donor T cells, mice transplanted with IDO1−/− pre-pDCs had increased GVHD-mortality.
Taken together, these data support a novel method of regulating the GVL and GVHD activity of donor T cells by selective manipulation of the content of donor DC precursors in the hematopoietic cell allograft. We propose a feedback model in which donor pDCs initially induce Th1 polarization of activated donor T cells that secrete high levels of IFN-γ. Local production of IFN-γ by donor CD4+ T cells induces IDO expression by donor pDCs that then initiate counterregulatory immune effects including the generation of T-regs and down-modulation of inflammatory cytokines in donor T cells, limiting their alloreactivity and limiting GVHD. Thus, a limited duration of alloreactivity in donor T cells in the host hematolymphoid microenvironment seems to be sufficient to mediate a durable GVL effect without leading to fatal GVHD.41 These findings have important implications for understanding the role of IDO and IDO inhibitors in clinical BMT and the treatment of GVHD post-BMT. Furthermore, novel strategies of graft engineering to modify the content or function of donor antigen-presenting cells may effectively regulate donor T-cell function to optimize the GVL activity of BMT in patients with hematologic malignancies.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dr David L. Jaye for histopathologic analysis of tissue sections from experimental animals, Dr Ying Wang for performing in vivo bioluminescent imaging, Dave Pinelli for assisting with competitive repopulation experiments to optimize the strategy for gating stem cells, and members of the Biostatistics Core at the Winship Cancer Institute for advice on statistical methods.
This work was supported by National Institutes of Health grant P01HL086773. Y.L. was supported by a grant from the Foundation for the author of National Excellent Doctoral Dissertation of China.
National Institutes of Health
Authorship
Contribution: Y.L. and C.R.G. designed and performed experiments, analyzed data, and wrote the manuscript; A.S. and J.M.L. designed and performed experiments and analyzed data; K.A.D. and L.M.O. designed and performed experiments; J.D.R. and J.G. helped design experiments and edited the paper; and E.K.W. designed experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Edmund K. Waller, Department of Hematology and Medical Oncology, Winship Cancer Institute, Room B5119, Emory University Medical School, Emory University, 1365B Clifton Rd NE, Atlanta, GA 30322; e-mail: ewaller@emory.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal