

TO THE EDITOR:
We have previously reported on the results of the United Kingdom National Cancer Research Institute (NCRI) AML17 trial, which, for acute promyelocytic leukemia (APL) of all risk groups, compared anthracycline + all-trans retinoic acid (ATRA) (AIDA) vs arsenic trioxide (ATO) + ATRA using an attenuated schedule of ATO.1 Here, we present long-term survival results for randomized patients and for 32 patients who received the same schedule of ATO + ATRA after relapsing from the AIDA arm. Ethics approval was provided by Wales REC 3, and all participants provided signed informed consent. From May 2009 to October 2013, 235 patients aged >16 years were randomized to ATRA (45 mg/m2 in 2 divided doses) + ATO (8 week induction [0.3 mg/kg on days 1-5 during week 1 and 0.25 mg/kg twice a week during weeks 2-8], followed by 4 consolidation courses of 0.3 mg/kg on days 1-5 during week 1 and 0.25 mg/kg twice a week during weeks 2-4 [63 ATO doses]) or the AIDA schedule: idarubicin (Ida) 12 mg/m2 on days 2, 4, 6, and 8 + ATRA to day 60 (induction) and then Ida 5 mg/m2 on days 1-4 + ATRA on days 1-15 (course 2), mitoxantrone 10 mg/m2 on days 1-5 + ATRA on days 1-15 (course 3), and Ida 12 mg/m2 on day l + ATRA on days 1-15 (course 4). ATRA was administered at 45 mg/m2 per day in 2 divided doses. Maintenance was not given. High-risk patients could receive a single dose of gemtuzumab ozogamicin (GO; day 1, 6 mg/m2). An additional 70 patients were treated with AIDA after closure of the randomization. In total, 189 patients were treated with AIDA, of whom 33 relapsed, and 32 were treated with the same schedule of ATO + ATRA, receiving a median of 4 cycles (range, 1-5). Follow-up is complete to 1 July 2017.
The median age was 47 years (range, 16-77); 57 had high-risk APL (white blood cells >10 × 109/L; 27 AIDA, 30 ATRA + ATO), and 49 were older than 60 years (24 AIDA, 25 ATRA + ATO). The results have not changed since our initial report1 : 91% entered morphological complete remission (CR), with no significant difference in CR rate between the arms (ATO + ATRA 94%, AIDA 89%; odds ratio [OR], 0.54; 95% confidence interval [CI], 0.21-1.34; P = .18). With a longer median follow-up of 67.4 months, the 5-year survival is now 92% (ATO + ATRA) vs 86% (AIDA) (HR, 0.71; 95% CI, 0.33-1.50; P = .4; Figure 1A). Among patients who became minimal residual disease negative after consolidation, molecular relapse-free survival (RFS) is 97% vs 78% (HR, 0.25; 95% CI, 0.12-0.52; P = .0002; Figure 1B). No patient treated with ATRA + ATO who became molecularly negative relapsed; among AIDA-treated patients, 5-year cumulative incidence of any relapse (including molecular) was 20%. A significant reduction in frank relapse (1% vs 10% at 5 years; hazard ratio [HR], 0.18; 95% CI, 0.05-0.60; P = .005) resulted in a better RFS for ATO (96% vs 86%; HR, 0.43; 95% CI, 0.18-1.03; P = .06; Figure 1C); the results are not affected by risk group (low risk 95% vs 87%; HR, 0.58; 95% CI, 0.22-1.55; P = .3; high risk 100% vs 83%; HR, 0.12; 95% CI, 0.02-0.84; P = .03; test for interaction P = .16). With additional follow-up, the molecular relapse risk with AIDA is higher than that observed in our previous AML15 trial (20% vs 9%) in which maintenance was used.2 However, the incidence of secondary acute myeloid leukemia/myelodysplastic syndrome was less in AML17 (1% vs 6%). No cases of secondary acute myeloid leukemia/myelodysplastic syndrome were seen after ATO + ATRA.
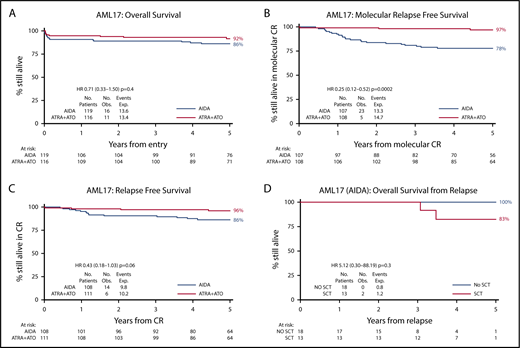
Clinical outcomes. (A) Overall survival for chemo-free vs AIDA approach. (B) Molecular RFS. (C) RFS. (D) Survival from relapse for relapsing AIDA patients treated with ATRA + ATO split by transplantation.
Clinical outcomes. (A) Overall survival for chemo-free vs AIDA approach. (B) Molecular RFS. (C) RFS. (D) Survival from relapse for relapsing AIDA patients treated with ATRA + ATO split by transplantation.
Of the 32 patients who relapsed following AIDA therapy, 1 died before treatment could be initiated, and 31 (5 with concomitant central nervous system [CNS] involvement) were treated with the same attenuated ATO schedule, including 17 at molecular relapse. Their 5-year overall survival is 88% with a median follow-up of 44.9 months. All 31 patients achieved molecular CR, of whom 5 patients received additional consolidation therapy with high-dose cytarabine (n = 4) or GO (n = 1) after achieving molecular remission. Thirteen patients were transplanted in CR2 (10 autograft, 3 allograft), including 4 of the 5 patients with CNS disease and the 5 patients who had received additional chemotherapy. Of the 18 patients treated with ATO + ATRA alone without transplant or consolidation chemotherapy, 14 remain in molecular remission, and 4 have relapsed (molecular in 3). Two patients (both transplanted) have died: 1 at 37 months postallograft in second molecular remission and the other at 36 months postautograft, who suffered a molecular relapse posttransplant. This patient had received only 1 course of ATO prior to autograft.
Genomic DNA was available from 31 patients who relapsed; paired diagnosis and hematological relapse samples were available from 8 patients, and diagnostic material only was available from a further 23 patients. We performed targeted next-generation sequencing of a panel of 60 genes frequently mutated in AML. Molecular barcoded libraries were prepared using the HaloPlexHS system and sequenced using an Illumina HiSeq 2500 instrument. Alignment and variant calling were performed using Agilent SureCall 4.0 software. In parallel, we performed polymerase chain reaction and fragment analysis to detect the FLT3 internal tandem duplication (ITD), as previously described.3 In keeping with previous reports, FLT3 ITD was the most frequent mutation and was detected in 14 of 31 patients (45%). Other recurrently mutated genes included WT1 in 7 patients (23%), NRAS in 3 patients (9.6%), and RUNX1 and KRAS in 2 patients each (6.4%). Profiling of the 5 patients who relapsed postsalvage ATO showed no consistent findings: 2 had FLT3 ITDs, 1 had NRAS/KRAS mutations, 1 had a WT1 mutation, and 1 did not have any mutations, suggesting that relapses post-ATO are not predictable based on upfront genomic data.
The primary purpose of this updated analysis was to establish whether an overall survival benefit for ATO + ATRA has emerged, as has been reported in the pivotal randomized APL0406 trial conducted in Italy and Germany by the Gruppo Italiano Malattie Ematologiche dell'Adulto (GIMEMA), AML Study Group, and Study Alliance Leukaemia cooperative groups.4,5 In AML17, although we found that the combination of ATO and ATRA continues to show a very low risk for relapse (1% at 5 years) irrespective of risk group, with a significantly superior RFS compared with AIDA, no overall survival benefit has emerged. These results are supported by a report from Abaza et al, who also observed a very low relapse risk in standard and high-risk APL patients treated with ATO + ATRA, supplemented by GO in high-risk patients.6 In that study, 46% of standard-risk patients also received GO for treatment of leukocytosis, an intervention that we used in only 6% of patients, suggesting that GO makes a minimal contribution to the low relapse risk with ATO, at least in standard-risk disease.
In AML17, the lack of survival benefit resulted from the highly effective salvage intervention for AIDA-treated patients with ATO, with the majority of patients treated at molecular relapse, which could be explained by the emphasis on minimal residual disease monitoring. For patients treated with first-line ATO and ATRA, molecular monitoring is now of questionable value once molecular CR has been documented due to the low risk for relapse; however, molecular surveillance remains important in those who have had ATO salvage for relapse. The alternative ATO dosing used in AML17 resulted in fewer days on ATO treatment and a correspondingly lower cumulative dose and drug acquisition costs than the GIMEMA-AML Study Group–Study Alliance Leukaemia schedule, despite the higher doses used in the first week of each course. This schedule proved highly effective as first-line therapy in standard-risk disease, as well as in high-risk patients and at relapse. These results also question the role of transplantation as consolidation for relapsed patients as has been recommended,7 at least in patients achieving molecular remission with ATO and ATRA who do not have CNS disease at relapse and who have received a full course of consolidation with ATO.
Acknowledgments
The authors thank Cancer Research UK for providing research support for the trial, Cephalon and Teva for providing ATO, the main investigators and colleagues who enrolled patients, Guy's and St Thomas' Hospital (London, UK) for curation of the molecular monitoring database, and the Haematology Clinical Trials Unit and Centre for Trials Research at Cardiff University.
This work was supported by Cardiff University.
Authorship
Contribution: A.B., N.R., R.H., and D.G. designed the trial; J.J., R.D., and D.G. performed molecular monitoring; S.B. collated the data; R.H. analyzed the data; N.R. and M.D. entered patients; and all authors contributed to the interpretation of the data and the writing of the manuscript. With the exception of D.G., all authors were able to undertake final review of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Alan Burnett is retired from the Cardiff University School of Medicine.
A complete list of members of the NCRI AML Working Group appears in “Appendix.”
Correspondence: Nigel Russell, Nottingham University Hospitals NHS Trust, A611 Hucknall Rd, Nottingham NG5 1PB, United Kingdom; e-mail: nigel.russell@nottingham.ac.uk.
Appendix: study group members
The members of the NCRI AML Working Group are: S. Ali, Castle Hill Hospital Hull, United Kingdom; S. Betteridge, Centre for Trials Research, Cardiff University, Cardiff, United Kingdom; K. Bowles, Norfolk and Norwich University Hospitals, Norwich, United Kingdom; D. Bowen, Leeds Teaching Hospitals NHS Trust, Leeds, United Kingdom; P. Cahalin, Blackpool Victoria Hospital, Blackpool, United Kingdom; J. Cavenagh, Barts NHS Trust, London, United Kingdom; R. Clark, Royal Liverpool University Hospital, Liverpool, United Kingdom; M. Copland, University of Glasgow, Glasgow, United Kingdom; C. Craddock, University Hospitals Birmingham, Birmingham, United Kingdom; D. Culligan, Aberdeen Royal Infirmary, Aberdeen, United Kingdom; M. Dennis, Christie Hospital, Manchester, United Kingdom; R. Dillon, Guy's Hospital, London, United Kingdom; S. Freeman, University of Birmingham, Birmingham, United Kingdom; R. Gale, University College London, London, United Kingdom; B. Gibson, Royal Hospital for Sick Children, Glasgow, United Kingdom; R. K. Hills, Centre for Trials Research, Cardiff University, Cardiff, United Kingdom; A. Hunter, Leicester Royal Infirmary, Leicester, United Kingdom; B. Huntly, University of Cambridge, Cambridge, United Kingdom; P. Johnson, Western General Hospital, Edinburgh, United Kingdom; G. Jones, Newcastle Hospitals, Newcastle upon Tyne, United Kingdom; H. Kaur, Royal Hallamshire Hospital, Sheffield, United Kingdom; J. Kell, University Hospital of Wales, Cardiff, United Kingdom; R. Kelly, Leeds Teaching Hospitals NHS Trust, Leeds, United Kingdom; A. Khwaja, University College Hospitals, London, United Kingdom; L. Kjeldsen, Rigshospitalet, Copenhagen, Denmark; S. Knapper, University Hospital of Wales, Cardiff, United Kingdom; P. Kottaridis, University College Hospitals, London, United Kingdom; R. Lown, Southampton General Hospital, Southampton, United Kingdom; M. F. McMullin, Queens University Belfast, Belfast, United Kingdom; P. Mehta, Bristol Royal Infirmary, Bristol, United Kingdom; K. Mills, Queens University Belfast, Belfast, United Kingdom; M. Nikolousis, University Hospitals Birmingham, Birmingham, United Kingdom; D. Richardson, Southampton General Hospital, Southampton, United Kingdom; N. Russell, Nottingham University Hospitals NHS Trust, Nottingham, United Kingdom; R. Spearing, Canterbury Hospitals, Canterbury, New Zealand; D. Taussig, Royal Marsden NHS Foundation Trust, London, United Kingdom; E. Tholouli, Manchester Royal Infirmary, Manchester, United Kingdom; I. Thomas, Centre for Trials Research, Cardiff University, Cardiff, United Kingdom; and P. Vyas, Oxford Radcliffe Hospitals NHS Trust, Oxford, United Kingdom.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal