

Key Points
Preemptive allogeneic stem cell transplantation improves prognosis of asymptomatic children genetically predisposed to primary HLH.
Abstract
Asymptomatic carriers (ACs) of pathogenic biallelic mutations in causative genes for primary hemophagocytic lymphohistiocytosis (HLH) are at high risk of developing life-threatening HLH, which requires allogeneic hematopoietic stem cell transplantation (HSCT) to be cured. There are no guidelines on the management of these asymptomatic patients. We analyzed the outcomes of pairs of index cases (ICs) and subsequently diagnosed asymptomatic family members carrying the same genetic defect. We collected data from 22 HSCT centers worldwide. Sixty-four children were evaluable. ICs presented with HLH at a median age of 16 months. Seven of 32 ICs died during first-line therapy, and 2 are alive after chemotherapy only. In all, 23/32 underwent HSCT, and 16 of them are alive. At a median follow-up of 36 months from diagnosis, 18/32 ICs are alive. Median age of ACs at diagnosis was 5 months. Ten of 32 ACs activated HLH while being observed, and all underwent HSCT: 6/10 are alive and in complete remission (CR). 22/32 ACs remained asymptomatic, and 6/22 have received no treatment and are in CR at a median follow-up of 39 months. Sixteen of 22 underwent preemptive HSCT: 15/16 are alive and in CR. Eight-year probability of overall survival (pOS) in ACs who did not have activated HLH was significantly higher than that in ICs (95% vs 45%; P = .02), and pOS in ACs receiving HSCT before disease activation was significantly higher than in ACs receiving HSCT after HLH activation (93% vs 64%; P = .03). Preemptive HSCT in ACs proved to be safe and should be considered.
Introduction
Primary hemophagocytic lymphohistiocytosis (HLH) is a rare disease with an estimated incidence of 1.8 per 100 000 live births per year.1,2 It is caused by a number of genetic mutations that affect the exocytosis of cytotoxic granules in T and NK cells, thus hampering their killing function.3 To date, 4 different genes have been identified as causative for primary HLH and an additional 5 genes are responsible for immunodeficiencies in which HLH is a prominent clinical feature (PRF1, STXBP2, UNC13D, STX11, RAB27A, BIRC4, SH2D1A, LYST, AP3B1).4-14 The curative treatment in primary HLH is hematopoietic stem cell transplantation (HSCT).15-21 Despite advances in chemoimmunotherapy for the treatment of primary HLH, overall survival (OS) remains at only about 59%, with virtually no survivors reported among patients who did not receive a transplant once the disease was fully active.22-24 Data from the largest cooperative prospective international studies (HLH-94 and HLH-2004 protocols) show that up to 20% of patients do not achieve a durable complete remission (CR) with first-line chemotherapy, and disease progression represents the overriding cause of death for patients not receiving HSCT within 12 months from disease onset.22 The initial HSCT experience with the use of myeloablative conditioning regimens was unsatisfactory.18,25,26 Since the introduction of reduced intensity conditioning (RIC), better results have been achieved with limited toxicities and OS of 75% to 92% for patients who do not have central nervous system (CNS) disease and who achieve good disease control before HSCT.20
Although HSCT is clearly the treatment of choice once HLH becomes clinically manifest, the question of whether to offer HSCT to asymptomatic siblings who have been diagnosed genetically before the onset of symptoms remains unanswered. To date there are no data on the natural history of asymptomatic siblings to guide clinicians or families in making this decision. The heterogeneity of clinical presentation in these autosomal recessive diseases renders decision-making extremely challenging. In fact, previous studies demonstrated that only one-third of relatives affected by the same genetic mutation as index cases (ICs) have a similar age at presentation.27 The risks of HSCT, often performed at a very young age, need to be balanced with the risk of waiting for the first HLH episode to manifest, which can be fatal in up to 20% of the patients.25 In this study, we describe for the first time the outcome of children who carry a genetic mutation predisposing to primary HLH and who are diagnosed while asymptomatic compared with that of ICs in the same family who have HLH that has activated.
Methods and data collection
Children younger than age 18 years who carried the same genetic mutations as a sibling affected by primary HLH and who were asymptomatic at the time of genetic diagnosis were eligible for this study. Only children diagnosed between 2005 and 2016 were eligible. Data were collected via a questionnaire distributed to centers through the Histiocyte Society. A key physician was identified to collect and report local data for each of the pediatric HSCT centers in Japan, Europe, and North America that participated. In brief, the questionnaire captured data on the clinical characteristics, management, and outcome of the ICs and of a subsequent asymptomatic family member with primary HLH (asymptomatic case [AC]). The questionnaire also asked participating centers about their current attitude toward the management of asymptomatic family members with primary HLH. Given the recognized variability of the disease course in patients with XLP1 and XIAP deficiency, these patients were not analyzed in this study. Patients enrolled in the study had given prior consent to the use of their anonymized clinical data in the context of national or international studies. Individual institutional review boards in North America approved this study.
Statistics
The results of descriptive analyses are reported as medians and ranges. Kaplan-Meier estimators and confidence intervals (CIs) are used to estimate OS, and the log-rank test and Fisher’s exact test (discrete variables) have been used to compare data. Patients were censored at last follow-up if no events occurred. CIs were reported at the 95% level, and statistical tests were performed at the 0.05 level (two sided).
Results
Eleven of 22 contacted centers submitted a total of 32 pairs or triplets of ICs and ACs corresponding to a total number of 33 ICs and 33 ACs. Because 2 patients were lost to follow-up, only 32 ICs and 32 ACs were evaluable for analysis. Seven centers had no patients fulfilling the inclusion criteria, and 4 centers did not reply to the questionnaire.
Patients’ characteristics and outcomes
Of the 32 pairs and triad of ICs and ACs identified, 13 had perforin (PRF1) deficiency, 6 had MUNC18-2 (STXBP2) deficiency, 4 had MUNC13-4 (UNC13D) deficiency, 2 had syntaxin (STX11) deficiency, 6 were diagnosed with Griscelli syndrome type II (RAB27A deficiency), and 1 was diagnosed with Chediak Higashi syndrome (LYST deficiency).
Their genetic and clinical characteristics are summarized in Table 1. For the whole cohort of patients, median age at HLH diagnosis was 9 months (range, 0 to 142 months), and median follow-up from diagnosis was 36 months (range, 1 to 144 months).
Genetic and clinical characteristics of the analyzed patients
| Patient | Protein deficit | Genetics | Age at diagnosis (mo) | Treatment | Disease status if AC at diagnosis | Outcome | Follow-up from diagnosis (mo) |
|---|---|---|---|---|---|---|---|
| IC 1 | MUNC18-2 | Compound heterozygous | 3 | Antithymocyte globulin + cyclosporine + steroids + HSCT | Died TRM | 5 | |
| AC 1 | MUNC18-2 | Compound heterozygous | 1 | HLH04 + HSCT | Activated during active follow-up | Alive CR | 22 |
| IC 2 | MUNC18-2 | 474-483delinsGA 1001C>T | 12 | Steroids | Alive CR | 48 | |
| AC 2 | MUNC18-2 | 474_483delinsGA 1001C>T | 24 | None | Never activated | Alive CR | 72 |
| IC 3 | MUNC18-2 | 1247G>C | 18 | HLH04 + rituximab + HSCT | Alive CR | 108 | |
| AC 3 | MUNC18-2 | 1247G>C | 60 | None | Never activated | Alive CR | 36 |
| IC 4 | MUNC18-2 | Unknown | 16 | HLH04 + HSCT | Died TRM | 72 | |
| AC 4 | MUNC18-2 | Unknown | 9 | HSCT | Never activated | Alive CR | 60 |
| IC 5 | MUNC18-2 | Compound heterozygous | NA | HLH04 + HSCT | Alive CR | 91 | |
| AC 5 | MUNC18-2 | Compound heterozygous | 6 | HSCT | Never activated | Alive CR | 75 |
| IC 6 | MUNC18-2 | 1247G>C | 138 | Steroids + intravenous immunoglobulin | Alive CR | 38 | |
| AC 6 | MUNC18-2 | 1247G>C | 108 | None | Never activated | Alive CR | 42 |
| IC 7 | LYST | 2749-50delAG | 24 | HLH04 + HSCT | Died TRM after graft loss and second HSCT | 26 | |
| AC 7 | LYST | 2749-50delAG | 102 | None | Never activated | Alive CR | 12 |
| IC 8 | PRF1 | 50delT 853-855delAAG | 5 | Unspecified chemotherapy | Died HLH progression | 1 | |
| AC 8 | PRF1 | 50delT 853-855delAAG | 5 | HLH04 + HSCT | Never activated; received prophylactic treatment pre-SCT | Alive CR | 32 |
| IC 9 | PRF1 | 445G>A 886T>C | 60 | Steroids | Died as a result of infection | 72 | |
| AC 9 | PRF1 | 445G>A 886T>C | 1 | HSCT | Never activated | Alive CR | 24 |
| IC 10 | PRF1 | Not available | 18 | Steroids + cyclosporine + alemtuzumab + HSCT | Alive CR | 12 | |
| AC 10 | PRF1 | Not available | 100 | Steroids + cyclosporine + alemtuzumab + HSCT | Activated while waiting for SCT | Alive CR | 12 |
| IC 11 | PRF1 | 254G>T;473C>T; 390C>T | 48 | HLH94 + HSCT | Alive CR | 36 | |
| AC 11 | PRF1 | 254G>T;473C>T; 390C>T | 9 | HSCT | Never activated | Alive CR | 38 |
| IC 12 | PRF1 | 1122G>A | 2 | HLH04 | Died HLH progression | 2 | |
| AC 12 | PRF1 | 1122G>A | Birth | HLH04 | Activated during prophylactic treatment while waiting for SCT | Lost to follow-up | |
| IC 13 | PRF1 | Not available | 4 | Etoposide + steroids + cyclosporine + intrathecal methotrexate + antithymocyte globulin + HSCT | Died HLH progression | 8 | |
| AC 13 | PRF1 | Not available | Prenatal | Steroids + cyclosporine + antithymocyte globulin + alemtuzumab + HSCT | Activated during prophylactic treatment while waiting for SCT | Died HLH progression | 15 |
| IC 14 | PRF1 | Not available | 54 | Steroids + cyclosporine + alemtuzumab + HSCT | Alive CR | 18 | |
| AC 14 | PRF1 | Not available | 75 | Steroids + cyclosporine + rituximab + HSCT | Activated during prophylactic treatment while waiting for SCT | Alive CR | 48 |
| IC 15 | PRF1 | 1376C>T | 36 | HLH04 + HSCT | Alive CR | 36 | |
| AC 15a | PRF1 | 1376C>T | 5 | HLH04 + HSCT | Activated while waiting for HSCT | Died TRM | 16 |
| AC 15b | PRF1 | 1376C>T | Prenatal | HSCT | Never activated | Alive CR | 17 |
| IC 16 | PRF1 | 50delT; 1034C | 8 | HLH94 + HSCT | Alive CR | 144 | |
| AC 16 | PRF1 | 50delT; 1034C | Birth | HLH94 + HSCT | Activated during active follow-up | Alive CR | 48 |
| IC 17 | PRF1 | 50delT; 1130G>A | 1 | Undefined induction + HSCT | Alive CR | 12 | |
| AC 17 | PRF1 | 50delT;1130G>A | Birth | HLH04 + HSCT | Activated during active follow-up | Alive CR | 62 |
| IC 18 | PRF1 | 1081A>T; 1081A>T | 60 | HLH04 + HSCT | Died TRM | 3 | |
| AC 18 | PRF1 | 1081A>T; 1081A>T | 30 | HSCT | Never activated | Alive CR | 21 |
| IC 19 | PRF1 | Not available | 72 | HLH04 + HSCT | Alive CR | 60 | |
| AC 19 | PRF1 | Not available | 3 | HLH04 + HSCT | Activated while waiting for HSCT | Alive CR | 72 |
| IC 20 | PRF1 | G5759C>T G5897A>C | 8 | HLH04 + HSCT | Alive CR | 36 | |
| AC 20 | PRF1 | G5759C>T G5897A>C | Prenatal | HSCT | Never activated | Alive CR | 18 |
| IC 21 | MUNC13-4 | 1389G>A; 1620-1621delCA | 1 | HLH04 + HSCT | Alive CR | 36 | |
| AC 21 | MUNC13-4 | 1389G>A; 1620-1621delCA | Prenatal | HSCT | Never activated | Alive CR | 60 |
| IC 22 | MUNC13-4 | 118-308C>T | 16 | Cyclosporine + etoposide (withdrew) | Died HLH progression | 2 | |
| AC 22 | MUNC13-4 | 118-308C>T | 13 | HSCT, lost graft, HLH04 + second HSCT | Never activated | Alive CR | 89 |
| IC 23 | MUNC13-4 | Not available | 2 | HLH94 + antithymocyte globulin + HSCT | Alive CR | 144 | |
| AC 23 | MUNC13-4 | Not available | 1 | None | Alive CR | 144 | |
| IC 24/a | MUNC13-4 | 118-308C>T; 1596+1G>C | 1 | HLH04 + HSCT | Alive CR | 45 | |
| IC 24/b | MUNC13-4 | 118-308C>T; 1596+1G>C | 1 | HLH04 + HSCT | Died TRM | 13 | |
| AC 24 | MUNC13-4 | 118-308C>T; 1596+1G>C | Birth | HSCT | Never activated | Alive CR | 26 |
| IC 25 | STX11 | 37+16>A | Birth | HLH04 | Died HLH progression | 1 | |
| AC 25 | STX11 | 37+16>A | Prenatal | Cyclosporine + steroids + HSCT | Never activated; receiving prophylactic treatment | Died; sudden death in CR | 17 |
| IC 26 | STX11 | Not available | 36 | HLH04 + HSCT | Alive CR | 120 | |
| AC 26 | STX11 | Not available | 72 | HLH94 + HSCT | Activated during active follow up | Died TRM | 76 |
| IC 27 | RAB27A | 281G>A | 18 | HLH04 + HSCT | Alive CR | 25 | |
| AC 27 | RAB27A | 281G> | 144 | None | Never activated | Alive CR | 2 |
| IC 28 | RAB27A | 467+1G>A | 12 | HLH94 | Lost to follow-up | NA | |
| AC 28 | RAB27A | 467+1G>A | Birth | Cyclosporine + HSCT | Never activated; receiving prophylactic treatment | Alive CR | 12 |
| IC 29 | RAB27A | 220G>C;335delA | 9 | Dexamethasone + etoposide | Died HLH progression | 36 | |
| AC 29 | RAB27A | 220G>C;335delA | 1 | HSCT | Never activated | Alive CR | 91 |
| IC 30 | RAB27A | Not available | 9 | Corticosteroids, cyclosporine, intrathecal methotrexate, HSCT × 3 | Alive CR | 84 | |
| AC 30 | RAB27A | Not available | Prenatal | Corticosteroids, cyclosporine, HSCT | Activated while waiting for HSCT | Died HLH progression | 18 |
| IC 31 | RAB27A | Not available | 126 | Alemtuzumab, corticosteroids, cyclosporine, intrathecal methotrexate, natalizumab, HSCT | Died TRM | 30 | |
| AC 31 | RAB27A | Not available | 180 | HSCT | Never activated; receiving prophylactic treatment while waiting for HSCT | Alive in CR | 36 |
| IC 32 | RAB27A | Not available | 84 | Cyclosporine + alemtuzumab + steroids | Died HLH progression | 12 | |
| AC 32 | RAB27A RAB27A | Not available | 84 | Cyclosporine + HSCT | Never activated; receiving prophylactic treatment while waiting for HSCT | Alive in CR | 26 |
| Patient | Protein deficit | Genetics | Age at diagnosis (mo) | Treatment | Disease status if AC at diagnosis | Outcome | Follow-up from diagnosis (mo) |
|---|---|---|---|---|---|---|---|
| IC 1 | MUNC18-2 | Compound heterozygous | 3 | Antithymocyte globulin + cyclosporine + steroids + HSCT | Died TRM | 5 | |
| AC 1 | MUNC18-2 | Compound heterozygous | 1 | HLH04 + HSCT | Activated during active follow-up | Alive CR | 22 |
| IC 2 | MUNC18-2 | 474-483delinsGA 1001C>T | 12 | Steroids | Alive CR | 48 | |
| AC 2 | MUNC18-2 | 474_483delinsGA 1001C>T | 24 | None | Never activated | Alive CR | 72 |
| IC 3 | MUNC18-2 | 1247G>C | 18 | HLH04 + rituximab + HSCT | Alive CR | 108 | |
| AC 3 | MUNC18-2 | 1247G>C | 60 | None | Never activated | Alive CR | 36 |
| IC 4 | MUNC18-2 | Unknown | 16 | HLH04 + HSCT | Died TRM | 72 | |
| AC 4 | MUNC18-2 | Unknown | 9 | HSCT | Never activated | Alive CR | 60 |
| IC 5 | MUNC18-2 | Compound heterozygous | NA | HLH04 + HSCT | Alive CR | 91 | |
| AC 5 | MUNC18-2 | Compound heterozygous | 6 | HSCT | Never activated | Alive CR | 75 |
| IC 6 | MUNC18-2 | 1247G>C | 138 | Steroids + intravenous immunoglobulin | Alive CR | 38 | |
| AC 6 | MUNC18-2 | 1247G>C | 108 | None | Never activated | Alive CR | 42 |
| IC 7 | LYST | 2749-50delAG | 24 | HLH04 + HSCT | Died TRM after graft loss and second HSCT | 26 | |
| AC 7 | LYST | 2749-50delAG | 102 | None | Never activated | Alive CR | 12 |
| IC 8 | PRF1 | 50delT 853-855delAAG | 5 | Unspecified chemotherapy | Died HLH progression | 1 | |
| AC 8 | PRF1 | 50delT 853-855delAAG | 5 | HLH04 + HSCT | Never activated; received prophylactic treatment pre-SCT | Alive CR | 32 |
| IC 9 | PRF1 | 445G>A 886T>C | 60 | Steroids | Died as a result of infection | 72 | |
| AC 9 | PRF1 | 445G>A 886T>C | 1 | HSCT | Never activated | Alive CR | 24 |
| IC 10 | PRF1 | Not available | 18 | Steroids + cyclosporine + alemtuzumab + HSCT | Alive CR | 12 | |
| AC 10 | PRF1 | Not available | 100 | Steroids + cyclosporine + alemtuzumab + HSCT | Activated while waiting for SCT | Alive CR | 12 |
| IC 11 | PRF1 | 254G>T;473C>T; 390C>T | 48 | HLH94 + HSCT | Alive CR | 36 | |
| AC 11 | PRF1 | 254G>T;473C>T; 390C>T | 9 | HSCT | Never activated | Alive CR | 38 |
| IC 12 | PRF1 | 1122G>A | 2 | HLH04 | Died HLH progression | 2 | |
| AC 12 | PRF1 | 1122G>A | Birth | HLH04 | Activated during prophylactic treatment while waiting for SCT | Lost to follow-up | |
| IC 13 | PRF1 | Not available | 4 | Etoposide + steroids + cyclosporine + intrathecal methotrexate + antithymocyte globulin + HSCT | Died HLH progression | 8 | |
| AC 13 | PRF1 | Not available | Prenatal | Steroids + cyclosporine + antithymocyte globulin + alemtuzumab + HSCT | Activated during prophylactic treatment while waiting for SCT | Died HLH progression | 15 |
| IC 14 | PRF1 | Not available | 54 | Steroids + cyclosporine + alemtuzumab + HSCT | Alive CR | 18 | |
| AC 14 | PRF1 | Not available | 75 | Steroids + cyclosporine + rituximab + HSCT | Activated during prophylactic treatment while waiting for SCT | Alive CR | 48 |
| IC 15 | PRF1 | 1376C>T | 36 | HLH04 + HSCT | Alive CR | 36 | |
| AC 15a | PRF1 | 1376C>T | 5 | HLH04 + HSCT | Activated while waiting for HSCT | Died TRM | 16 |
| AC 15b | PRF1 | 1376C>T | Prenatal | HSCT | Never activated | Alive CR | 17 |
| IC 16 | PRF1 | 50delT; 1034C | 8 | HLH94 + HSCT | Alive CR | 144 | |
| AC 16 | PRF1 | 50delT; 1034C | Birth | HLH94 + HSCT | Activated during active follow-up | Alive CR | 48 |
| IC 17 | PRF1 | 50delT; 1130G>A | 1 | Undefined induction + HSCT | Alive CR | 12 | |
| AC 17 | PRF1 | 50delT;1130G>A | Birth | HLH04 + HSCT | Activated during active follow-up | Alive CR | 62 |
| IC 18 | PRF1 | 1081A>T; 1081A>T | 60 | HLH04 + HSCT | Died TRM | 3 | |
| AC 18 | PRF1 | 1081A>T; 1081A>T | 30 | HSCT | Never activated | Alive CR | 21 |
| IC 19 | PRF1 | Not available | 72 | HLH04 + HSCT | Alive CR | 60 | |
| AC 19 | PRF1 | Not available | 3 | HLH04 + HSCT | Activated while waiting for HSCT | Alive CR | 72 |
| IC 20 | PRF1 | G5759C>T G5897A>C | 8 | HLH04 + HSCT | Alive CR | 36 | |
| AC 20 | PRF1 | G5759C>T G5897A>C | Prenatal | HSCT | Never activated | Alive CR | 18 |
| IC 21 | MUNC13-4 | 1389G>A; 1620-1621delCA | 1 | HLH04 + HSCT | Alive CR | 36 | |
| AC 21 | MUNC13-4 | 1389G>A; 1620-1621delCA | Prenatal | HSCT | Never activated | Alive CR | 60 |
| IC 22 | MUNC13-4 | 118-308C>T | 16 | Cyclosporine + etoposide (withdrew) | Died HLH progression | 2 | |
| AC 22 | MUNC13-4 | 118-308C>T | 13 | HSCT, lost graft, HLH04 + second HSCT | Never activated | Alive CR | 89 |
| IC 23 | MUNC13-4 | Not available | 2 | HLH94 + antithymocyte globulin + HSCT | Alive CR | 144 | |
| AC 23 | MUNC13-4 | Not available | 1 | None | Alive CR | 144 | |
| IC 24/a | MUNC13-4 | 118-308C>T; 1596+1G>C | 1 | HLH04 + HSCT | Alive CR | 45 | |
| IC 24/b | MUNC13-4 | 118-308C>T; 1596+1G>C | 1 | HLH04 + HSCT | Died TRM | 13 | |
| AC 24 | MUNC13-4 | 118-308C>T; 1596+1G>C | Birth | HSCT | Never activated | Alive CR | 26 |
| IC 25 | STX11 | 37+16>A | Birth | HLH04 | Died HLH progression | 1 | |
| AC 25 | STX11 | 37+16>A | Prenatal | Cyclosporine + steroids + HSCT | Never activated; receiving prophylactic treatment | Died; sudden death in CR | 17 |
| IC 26 | STX11 | Not available | 36 | HLH04 + HSCT | Alive CR | 120 | |
| AC 26 | STX11 | Not available | 72 | HLH94 + HSCT | Activated during active follow up | Died TRM | 76 |
| IC 27 | RAB27A | 281G>A | 18 | HLH04 + HSCT | Alive CR | 25 | |
| AC 27 | RAB27A | 281G> | 144 | None | Never activated | Alive CR | 2 |
| IC 28 | RAB27A | 467+1G>A | 12 | HLH94 | Lost to follow-up | NA | |
| AC 28 | RAB27A | 467+1G>A | Birth | Cyclosporine + HSCT | Never activated; receiving prophylactic treatment | Alive CR | 12 |
| IC 29 | RAB27A | 220G>C;335delA | 9 | Dexamethasone + etoposide | Died HLH progression | 36 | |
| AC 29 | RAB27A | 220G>C;335delA | 1 | HSCT | Never activated | Alive CR | 91 |
| IC 30 | RAB27A | Not available | 9 | Corticosteroids, cyclosporine, intrathecal methotrexate, HSCT × 3 | Alive CR | 84 | |
| AC 30 | RAB27A | Not available | Prenatal | Corticosteroids, cyclosporine, HSCT | Activated while waiting for HSCT | Died HLH progression | 18 |
| IC 31 | RAB27A | Not available | 126 | Alemtuzumab, corticosteroids, cyclosporine, intrathecal methotrexate, natalizumab, HSCT | Died TRM | 30 | |
| AC 31 | RAB27A | Not available | 180 | HSCT | Never activated; receiving prophylactic treatment while waiting for HSCT | Alive in CR | 36 |
| IC 32 | RAB27A | Not available | 84 | Cyclosporine + alemtuzumab + steroids | Died HLH progression | 12 | |
| AC 32 | RAB27A RAB27A | Not available | 84 | Cyclosporine + HSCT | Never activated; receiving prophylactic treatment while waiting for HSCT | Alive in CR | 26 |
HLH04, treatment according to the HLH04 protocol from the International Histiocyte Society; SCT, stem cell transplantation.
Of the 64 total evaluable patients, 45 were alive; thus the probability of OS (pOS) in the whole cohort of patients was 56% (95% CI, 35%-70%) (Figure 1A). Forty-nine patients underwent HSCT, 8 of 49 died as a result of transplantation-related mortality (TRM), and 3 died as a result of post-HSCT HLH progression. One patient had an unexplained sudden death while in CR at 12 months after HSCT. Thirty-seven of 49 are alive at a median follow-up of 36 months from diagnosis (8-year pOS for HSCT patients, 63%; 95% CI, 38%-77%). Fifteen patients did not receive HSCT, 1 died as a result of complications related to first-line treatment, and 6 died as a result of HLH progression. Eight of those patients are alive at a median follow-up of 12 months (pOS for non-HSCT patients, 41%; 95% CI, 7%-74%).
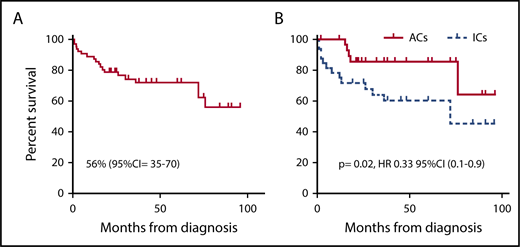
OS per Kaplan-Meier estimate comparing ICs and ACs. (A) The whole population of patients; (B) our population of patients. HR, hazard ratio.
OS per Kaplan-Meier estimate comparing ICs and ACs. (A) The whole population of patients; (B) our population of patients. HR, hazard ratio.
ICs
Per inclusion criteria, all ICs (n = 32) (Figure 2) had overt disease at the time of diagnosis. The median age of the ICs at disease presentation was 16 months (range, 0 to 138 months). Median follow-up for the ICs was 36 months from diagnosis (range, 1-144 months). Fourteen of 32 had CNS involvement, and 6 were refractory to first-line treatment.
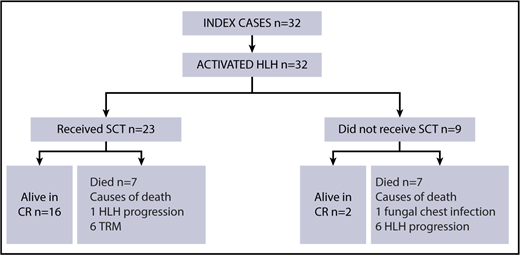
Seven patients died during first-line therapy (1 as a result of treatment-related toxicity, 6 as a result of HLH progression) and did not survive to HSCT. Twenty-three of 32 patients underwent HSCT. Six died as a result of TRM (1 as a result of chronic graft-versus-host disease, 1 as a result of lymphoproliferative disease and gram-negative sepsis, 1 as a result of infection following graft loss and second HSCT, 1 as a result of gram-negative sepsis, and 2 as a result of unspecified TRM) and 1 died as a result of disease progression after haploidentical HSCT. Sixteen children are alive and in CR after HSCT. Altogether, 18 of 32 evaluable ICs were alive at the latest follow-up (8-year pOS for ICs, 45%; 95% CI, 21%-62%). Two patients affected by MUNC18-2 deficiency did not receive HSCT and were alive at a median follow-up of 42 months from diagnosis (for details, see ICs 2 and 6 in supplemental Table 1 available on the Blood Web site).
ACs
Among the ACs (n = 32) (Figure 3), 10 (31%) of 32 had activated HLH during observation (n = 4) or while waiting for HSCT (n = 6); 3 were receiving cyclosporine at the time of HLH activation. Of these 10 patients, 1 had CNS disease, and 3 required multiple lines of treatment to achieve remission. All 10 patients underwent HSCT: 6 are alive in CR and 4 died (2 as a result of disease progression and 2 as a result of TRM).

Of the 22 patients who did not experience activated HLH during observation, 5 were receiving prophylactic treatment while waiting for HSCT. Sixteen of 22 underwent HSCT as a preemptive measure; 15 are alive in CR, 1 had a sudden death as a result of unknown causes at 19 months of age and at 12 months after HSCT while in CR. Six of 22 patients (3 with MUNC18-2 deficiency, 1 with LYST deficiency, 1 with RAB27A deficiency, and 1 with MUNC13-4 deficiency) have received no specific treatment and are alive and are in CR at a median follow-up of 39 months from diagnosis. These 6 children are now older than their siblings were at the time of disease presentation; in 2 cases the corresponding ICs have been treated with chemotherapy only, and they achieved a good degree of disease remission and did not require HSCT (AC2, -3, -6, -7, -23, and -27; Table 1). Overall 21 of 22 ACs without HLH activation are alive with no survival difference between those who underwent SCT and those who were actively followed up (93% vs 100%; P = 1).
Of 32 evaluable ACs, 27 were alive at last follow-up (8-year pOS, 63%; 95% CI, 20%-87%). Twenty-one of 26 patients who underwent HSCT are alive. Importantly, 15 of 16 patients who received a transplant before the development of HLH are alive compared with 6 of 10 who received a transplant after the development of HLH (pOS, 64% vs 93%; P = .032; hazard ratio [HR], 0.13; 95% CI, 0.02-0.84).
Impact of genetic abnormality on OS
None of the genetic subgroups had a statistically significant difference in survival compared with the pOS of 56% for the whole group. Of evaluable patients, 19 of 26 with perforin deficiency were alive as well as 1 of 4 with syntaxin 11 deficiency, 7 of 9 with MUNC13-4 deficiency, 10 of 12 with MUNC18-2 deficiency, 7 of 11 with Griscelli type 2 syndrome, and 1 of 2 with Chédiak-Higashi syndrome.
Concordance between sibling pairs with activated HLH disease
Nine pairs in which the AC experienced activated HLH were analyzed for concordance in presenting features. Seven pairs had perforin deficiency, 1 pair had syntaxin 11 deficiency, and 1 pair had MUNC18-2 deficiency. Age at disease activation was variable, with a difference between ICs and ACs of 2 weeks to 7 years. In 6 pairs, CNS status at diagnosis was discordant. Epstein-Barr virus (EBV) infection was a trigger for HLH in the IC with syntaxin 11 deficiency but was not involved in the disease activation of her sibling.
Discussion
HSCT is the standard treatment for primary HLH. However, ∼30% of patients die before HSCT because of uncontrolled HLH or treatment-related toxicity; hence, survival in children presenting with primary HLH is poor at 59%.22,24 Children who have well-controlled disease and survive HSCT have a superior survival (pOS, 71%).20,22,24 However, the decision to offer HSCT to ACs before the onset of HLH is complicated by the interplay between genetic and environmental factors (typically infectious triggers) that lead to the onset of HLH, incomplete genotype-phenotype correlation,10,27-30 and the ethical aspects of subjecting a young asymptomatic child to a toxic procedure with a significant TRM.
Consistent with the findings from the HLH-94 and HLH-2004 studies,22,23 ICs in our study had an OS of ∼50%: 7 of 32 ICs died before HSCT, whereas the incidence of TRM was 26%. Survival was significantly improved in ACs compared with ICs, irrespective of treatment received (63% vs 45%; P = .02) (Figure 1B). The majority of ACs received a preemptive transplant, and patients treated with this approach showed a survival of 15 (93%) of 16; the only death in this group was a result of causes not related to transplantation. Although numbers are small, it is interesting to note that survival in ACs who experienced HLH activation before HSCT was not statistically different from that of ICs (33% vs 45%; P = .9; 95% CI, 0.3-2.7; Figure 4). In contrast, ACs who received a preemptive transplant showed an improved survival compared with ACs who received a transplant once HLH had developed (pOS, 93% vs 64%; P = .03; HR, 0.13; 95% CI, 0.02-0.8).
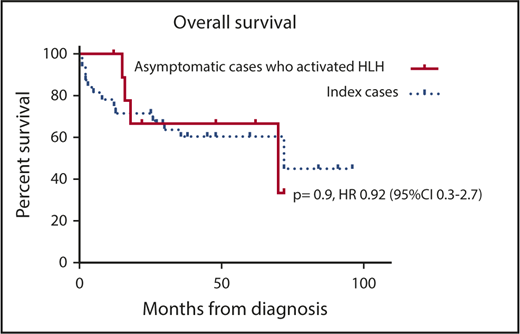
OS per Kaplan-Meier estimate in our population of patients comparing ACs who did not experience activated HLH with those who experienced activated HLH while on follow-up.
OS per Kaplan-Meier estimate in our population of patients comparing ACs who did not experience activated HLH with those who experienced activated HLH while on follow-up.
The long-term outcome of 6 of 20 ACs who did not receive a transplant remains to be determined. Median follow-up for this subgroup was 39 months, and at the latest follow-up, they were all older than their corresponding ICs at the time of disease onset. Although Cetica et al27 reported wide variation in the age of the IC at HLH onset and the ages of subsequent siblings in 9 of 26 familial cases, all eventually developed HLH except 1 who remained asymptomatic until the age of 25 years. The mutation profile of these patients can only partially explain why they remained asymptomatic. Indeed, although AC 3 and 6 had an MUNC18-2 mutation predicted to have a mild disease course,30 this cannot be documented for AC 2; moreover, AC 7, 23, and 27 were affected by LYST, MUNC13-4, and RAB27A deficiency, respectively, in which no genotype-phenotype correlation has been described. At present, the prognosis for this group is guarded at best.
In our cohort of patients, the tightest correlation between ICs and ACs in terms of disease onset was in the PRF1-deficient group. This group was also more likely to experience activated HLH early in life; 7 of 11 of the ACs with PRF1 deficiency experienced activated full-blown disease at a median time of 1 month from their diagnosis. Although it is not possible to make firm conclusions because the number of patients is too small, most patients in this group had mutations that would predict for absent perforin expression. Three patients had a combination of del50T mutation with other mutations, 2 were documented to have absent perforin expression by flow cytometry,29 and 1 patient was homozygous for c.1122G>A mutation, which has been described as being related to a severe and early phenotype of the disease (supplemental Table 1).10 Three other patients had no genetic data available for consideration, but one of them had absent perforin expression on cytometric analysis. These observations match the reported data on patients with biallelic disruptive mutations in the PRF1 gene who develop HLH at a significantly younger age compared with patients with missense mutations only.28 Preemptive transplantation might therefore be particularly indicated for this subgroups of patients.
At the other end of the spectrum, patients in our cohort with MUNC18-2 deficiency exhibited a milder form of HLH. Only 1 of 5 ACs experienced activated HLH and had a successful HSCT. Among the other 5 ACs with MUNC18-2 deficiency, 2 received preemptive HSCTs, and 3 are being actively followed up. In addition, 2 IVs with MUNC18-2 deficiency are alive without HSCT at a median of 44 months of follow-up from diagnosis. Among patients with MUNC18-2 deficiency who did not receive treatment and who are being actively observed with no signs of disease, 3 had homozygous mutation 1247-1G>C previously described as being associated with a mild phenotype postulated to result from expression of an abnormal protein.30 Ectopic expression of wild-type MUNC18-2 overcoming the MUNC18-2 deficiency was also suggested as a possible explanation for the milder HLH phenotype in some patients with MUNC18-2 deficiencies.13 We could not prove this phenomenon in our patients, but our report confirms a possible milder type of disease. The complexity of the clinical phenotype in MUNC18-2 deficiency, even in the presence of null mutations and the variability of multiorgan involvement, emphasizes the need for further studies and long-term follow-up in this and other HLH subtypes.
TRM in our case series was acceptable, with an incidence of 8 (16%) of 49, and we documented no significant difference in TRM between ICs and ACs who had activated HLH (26% vs 20%; P = 1.00). Significantly, no ACs died before HSCT, including those in the group who experienced activated HLH. We can speculate that ACs received HSCT earlier than ICs once they were diagnosed, and it was therefore easier to control their disease, but our data are not detailed enough to confirm this finding. Despite small numbers of patients, it is important to note that there was no TRM in the 16 ACs who received a transplant before they developed HLH. Although our study did not address the impact of donor choice or disease status on HSCT outcome, it seems that the best HSCT results were seen in ACs who received a preemptive transplant. A preemptive HSCT approach has been documented to be efficacious in the context of other primary immunodeficiencies,31 which allows for significant improvement in overall outcome. Our data strongly suggest that most ACs with HLH should receive a transplant before the onset of symptoms if an adequate donor is available.
In summary, our data confirm that primary HLH can have an unpredictable and severe course, even in ACs who are being carefully monitored. The initial presentation may involve the CNS and be refractory to primary therapy. Once HLH develops, the OS is not different from that of ICs. Preemptive HSCT before the onset of HLH could offer a survival advantage, especially in the subgroups of patients with a genotype predictive of severe disease, such as in children with a complete lack of perforin expression.28 Conversely, patients with an MUNC18-2 deficiency might require further consideration before HSCT because of the potential for milder forms of the disease and because of the possibility of multiorgan involvement that may not be cured by HSCT. On the basis of our data, we would recommend preemptive HSCT in ACs with certain subtypes of primary HLH (ie, PRF1 mutations with absent perforin expression and RAB27A mutation). In those ACs who have other genetically confirmed primary HLH mutations with unclear genotype-phenotype correlation, we would encourage considering HSCT in the presence of a well-matched donor after adequate counseling of families. A watch-and-wait policy could be suggested for patients affected with MUNC18-2 deficiency with mutations associated with milder phenotypes. Longer follow-up and larger patient numbers are needed to make more accurate genotype-phenotype correlations, which may in turn allow for more tailored treatment guidance on specific subgroups of patients.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Histiocyte Society for supporting this study and helping to deliver the questionnaire for data collection to pediatric hematopoietic stem cell transplantatio centers worldwide, and Geneviève de Saint Basile for her diagnostic contribution to the French cohort of patients.
Authorship
Contribution: G.L. analyzed the data and wrote the paper; R.M., P.V., and K.R. designed the study and critically reviewed the collected data; and all other authors contributed clinical data, commented on the preliminary results of the study, and suggested further analyses for the betterment of the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Kanchan Rao, Great Ormond Street Hospital, Great Ormond St, London WC1N 3JH, United Kingdom; e-mail: kanchan.ral@gosh.nhs.uk.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal