

Key Points
Dependencies on BCL2, BCLXL, MCL1 of primary myeloma cells defined using a BH3-mimetic toolkit differ between diagnosis and relapse.
Disruption of BAK/MCL1 complexes is crucial for apoptosis induced by the MCL1 mimetic; BCLXL is the main factor of resistance in myeloma.

Abstract
BH3 mimetics are promising drugs for hematologic malignancies that trigger cell death by promoting the release of proapoptotic BCL2 family members from antiapoptotic proteins. Multiple myeloma is considered to be a disease dependent mainly on MCL1 for survival, based mostly on studies using cell lines. We used a BH3-mimetic toolkit to study the dependency on BCL2, BCLXL, or MCL1 in malignant plasma cells from 60 patients. Dependencies were analyzed using an unbiased BH3 mimetics cell-death clustering by k-means. In the whole cohort of patients, BCL2 dependency was mostly found in the CCND1 subgroup (83%). Of note, MCL1 dependence significantly increased from 33% at diagnosis to 69% at relapse, suggesting a plasticity of the cellular dependency favoring MCL1 dependencies at relapse. In addition, 35% of overall patient samples showed codependencies on either BCL2/MCL1 or BCLXL/MCL1. Finally, we identified a group of patients not targeted by any of the BH3 mimetics, predominantly at diagnosis in patients not presenting the common recurrent translocations. Mechanistically, we demonstrated that BAK is crucial for cell death induced by MCL1 mimetic A1210477, according to the protection from cell death observed by BAK knock-down, as well as the complete and early disruption of MCL1/BAK complexes on A1210477 treatment. Interestingly, this complex was also dissociated in A1210477-resistant cells, but free BAK was simultaneously recaptured by BCLXL, supporting the role of BCLXL in A1210477 resistance. In conclusion, our study opens the way to rationally use venetoclax and/or MCL1 BH3 mimetics for clinical evaluation in myeloma at both diagnosis and relapse.
Introduction
Apoptotic deregulation is a hallmark of cancer cells. Interactions between the proteins of the BCL2 family play a pivotal role in the control of the intrinsic pathway of apoptosis. BCL2 family proteins encompass antiapoptotic members (BCL2, MCL1, and BCLXL), proapoptotic effectors (BAX and BAK), and proapoptotic BH3-only; among the latter are the BH3-only direct activators BIM, BID, and PUMA and sensitizers/repressors such as NOXA, BAD, BIK, BMF, and HRK.1 Antiapoptotic proteins exert their survival function by directly binding and inhibiting the function of proapoptotic BH3-only proteins and proapoptotic effectors, which can be present in a constitutive active state.1,2 Despite major advances in the understanding of the mitochondrial apoptotic pathway, many challenges remain to achieving its best possible exploitation in cancer treatment; notably, the accurate identification of tumor cell dependency on individual antiapoptotic family members. The reliability of the different approaches used to determine dependency, and their feasibility of application in preclinical studies, remain questions of debate.
Multiple myeloma (MM) is a cancer of plasma cells displaying a molecular heterogeneity, which includes hyperdiploid patients and patients with a translocation of the immunoglobulin (Ig) H locus on chromosome 14 with different chromosomes (4, 11, 6, or 16), leading to an overexpression of MMSET, CCND1, CCND3, or MAF genes, respectively.3 Despite the introduction of new drugs in the treatment of MM, which have substantially improved the overall survival, this malignancy remains incurable.4 We pinpoint that MM subgroups are heterogeneous for antiapoptotic member expression, and that the combined profile of BCL2, MCL1, and BCLXL discriminates the different MM molecular groups.5 MCL1 is frequently overexpressed either by gene amplification (1q amplification) or by oncogenic pathways.6 In MM, the amplification of 1q was shown to be associated with a poor prognosis.7 To pharmacologically inhibit MCL1, different selective MCL1 inhibitors have been developed; A1210477, the first inhibitor described, induces apoptosis in a mechanistically identical manner to MCL1 gene silencing in breast cancer or non-small cell lung cell lines.8,9 Very recently, S63845 has been identified as a very potent and selective MCL1 inhibitor, able to kill MCL1-dependent cell lines. It demonstrated a potent in vivo efficacy in preclinical xenograft mouse models of myeloma and lymphoma.10 Furthermore, we and others have demonstrated that venetoclax (ABT-199), the first clinically available BH3 mimetic specifically targeting BCL2, was particularly efficient in t(11;14) cell lines and primary MM samples expressing high BCL2 and low MCL1 and BCLXL levels.11-14 From a clinical perspective, the fact that BH3 mimetics targeting either BCL2 or MCL1 are now available gives rise to an urgent need to better define the cellular dependencies not only of MM cell lines but, more important, of primary MM cells. Until now, only 1 study has evaluated the cellular dependencies of primary cells using the BH3 profiling approach in a modest cohort of patients with myeloma.15 In the present study, we used a BH3-mimetic toolkit to define cellular dependencies on prosurvival BCL2 family proteins in primary MM cells (n = 60). Furthermore, we decipher more precisely the mechanism of action of the MCL1 BH3 mimetic, aiming to clarify the players that sustain MCL1 dependency and the potential factor of resistance.
Methods
Human myeloma cell lines and primary myeloma cells
Human myeloma cell lines (HMCLs) were characterized as previously described.16 After informed consent, MM bone marrow/blood samples were collected at University Hospital of Nantes.
Cell death assays in HMCLs and primary cells
Cell death in HMCLs was determined by Annexin V-fluorescein isothiocyanate staining. Cell death assay of primary myeloma cells was performed using mononuclear cells cultured in RPMI-1640 media with 5% fetal calf serum, 3 ng/mL interleukin 6 (IL-6) with/without the specific BH3 mimetic. Venetoclax, A1155463, and A1331852 were tested at 100, 300, and 1000 nM; A1210477 at 2.5 and 5 μM. Cell death was routinely measured by the loss of CD138 staining, as previously described.17 Alternatively, cell death was confirmed by Annexin V staining in CD38high myeloma cells.
Immunoblotting and MCL1 ubiquitination
BH3-profiling and cytochrome c release
BH3-profiling using MS1/Noxa peptide was performed as previously described.20 For cytochrome c release, cells were treated or not with A1210477.
Results
Dissecting BCL2, BCLXL, and MCL1 dependence in primary myeloma cells
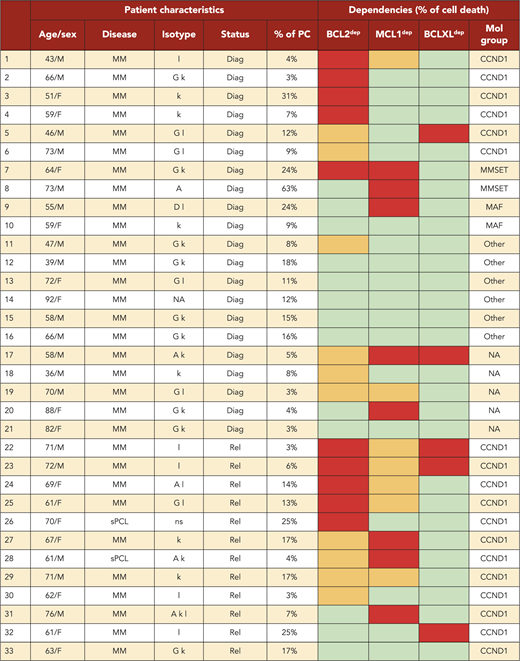
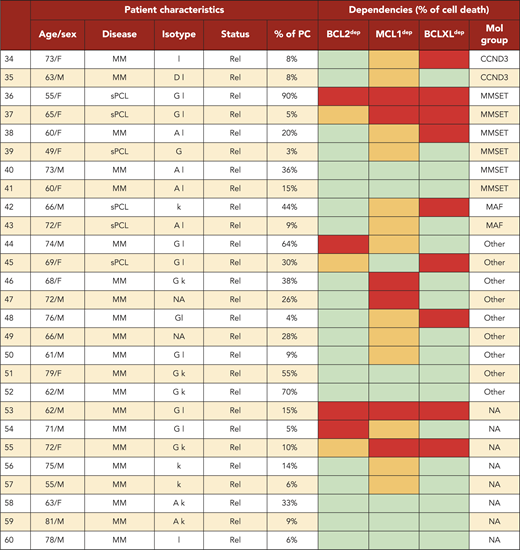
A BH3-mimetic toolkit was used to determine BCL2, BCLXL, and MCL1 dependence by ex vivo treatment of mononuclear cells from primary myeloma samples (Figure 1A). We analyzed the cellular dependence on individual antiapoptotic BCL2 members of 60 consecutive myeloma samples with a percentage of plasma cells of at least 3%. Patients were distributed as follows: 21 at diagnosis and 39 at relapse, including 7 secondary plasma cell leukemia (sPCL). Mononuclear cells from patients were treated with the respective BH3 mimetic overnight at the following concentrations: 100, 300, and 1000 nM for BCL2 and BCLXL BH3 mimetics; A1210477 MCL1 BH3 mimetic was used at 2.5 and 5 μM. Apoptosis was assessed by the loss of CD138 expression, as previously described,17 and confirmed by Annexin V staining (supplemental Figure 1). To define dependency groups (high, intermediate, and nondependent) in an unbiased way, cell death clustering by k-means was performed as described in supplemental Methods.21 When data were missing, data imputation for cell death was assessed by Multiple Imputation with Principal Component Analysis (MIPCA) and considered reliable,22 allowing continued clustering (supplemental Figure 2A-B). Cell death clustering by k-means retrieved an optimal number of 3 clusters for both BCL2 and MCL1 BH3 mimetics, whereas 2 was the optimal number of clusters for the BCLXL BH3 mimetic (Figure 1B; Table 1). Thus, at diagnosis, we first observed that 52% of primary samples were BCL2 dependent, whereas only 10% were BCLXL dependent. The dependence on BCL2 (either high or intermediate) and BCLXL was not significantly different between diagnosis and relapse stage (Figure 2A). Of note, only 1 of 14 samples was exclusively dependent on BCLXL (Figure 2C). Strikingly, we found that the MCL1 dependency was 33% at diagnosis, and increased to 69% at relapse, indicating a significant increase in MCL1 dependency during disease progression (P = .01; Figure 2A). Of note, the increase in MCL1 dependency was exclusively a result of the increase in the intermediate MCL1 dependency (P = .004; Figure 2B). The last 20 samples of our cohort were also investigated for S63845 sensitivity, demonstrating a strong correlation between A1210477 and S63845 responses (r = 0.78; P = .0001; supplemental Figure 3A). This indicates that both MCL1 BH3 mimetics could be used to determine MCL1 dependency. Incidentally, we also report the case of a patient at relapse who had been evaluated for dependencies at 2 different points (1-year interval; supplemental Figure 3B). The venetoclax response decreased from 28% to 1% of cell death, whereas A1210477 response increased from 76% to 90%, suggesting a plasticity of BCL2 and MCL1 dependencies (supplemental Figure 3B). In addition, we identified primary MM cells that did not depend on any of the 3 prosurvival molecules not only at diagnosis (33%) but also at relapse (20%). Moreover, codependencies were observed at both diagnosis (24%) and relapse (46%; Figure 2C). Among the 60 patients analyzed for cell dependencies, biological material from 47 patients was available for further analyses. Thus, 32 patients were assigned to the following molecular subgroups: CCND1, CCND3, MMSET, MAF (Table 1; supplemental Table 1). The 15 patient samples not harboring the abovementioned recurrent translocations were classified as “Others.” Cell dependencies were then analyzed in the different molecular groups of patients (n = 47; Figure 2D-F).

Dependence of primary MM samples on antiapoptotic BCL2 molecules. (A) Selectivity of BH3 mimetics for the respective antiapoptotic protein. The binding affinity of each compound for BCL2 antiapoptotic proteins was previously described.8,36,37 (B) Data clustering as assessed by k-means is displayed for BCL2, MCL1, and BCLXL BH3 mimetics (n = 1000 initiations of algorithm); values indicate Pearson correlation coefficients for the considered doses of the respective dependency group. Patient dependencies were defined as red, high; orange, intermediate; green, not dependent, and detailed in Table 1.
Dependence of primary MM samples on antiapoptotic BCL2 molecules. (A) Selectivity of BH3 mimetics for the respective antiapoptotic protein. The binding affinity of each compound for BCL2 antiapoptotic proteins was previously described.8,36,37 (B) Data clustering as assessed by k-means is displayed for BCL2, MCL1, and BCLXL BH3 mimetics (n = 1000 initiations of algorithm); values indicate Pearson correlation coefficients for the considered doses of the respective dependency group. Patient dependencies were defined as red, high; orange, intermediate; green, not dependent, and detailed in Table 1.
Ex vivo sensitivity of primary myeloma cells to BH3 mimetics
  |
|
BCL2, BCLXL, and MCL1 dependencies were defined as indicated in Figure 1B (red, high; orange, intermediate, green, not dependent). Molecular groups were determined either by fluorescence in situ hybridization or by quantitative polymerase chain reaction gene expression of anchorage genes of each molecular subgroup in purified CD138+ myeloma cells as defined in supplemental Table 1. Diag, diagnosis; F, female; M, male; NA, data not available; ns, nonsecreting; PC, plasma cells; Rel, relapse.
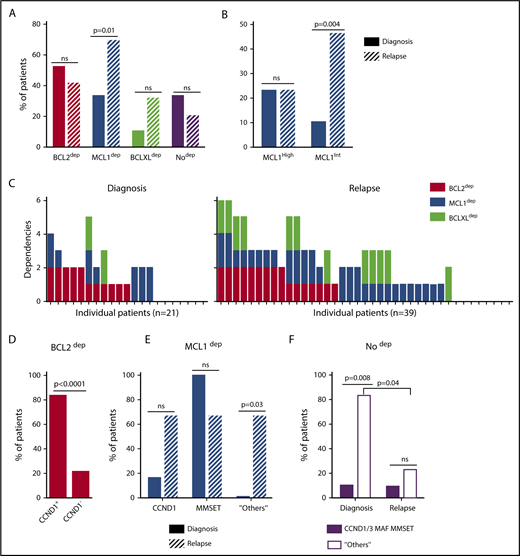
Dependence of primary MM samples on antiapoptotic BCL2 molecules, according to molecular subgroups. (A) Analyses of BCL2, MCL1, or BCLXL dependencies of 60 patients with MM at diagnosis and relapse. Patients’ dependencies to BCL2, MCL1, or BCLXL were defined by cell death response to the respective BH3 mimetic in primary myeloma cells, as indicated in Figure 1B and Table 1. Nodep includes patient samples that are insensitive to each 1 of the 3 BH3 mimetics (Table 1). Fisher’s exact test was used for statistical analysis. (B) High and intermediate patients’ dependencies to MCL1 were analyzed at diagnostic versus relapse. Fisher’s exact test was used for statistical analysis. (C) Analyses of individual dependencies of 60 patients with MM at diagnosis and relapse. For each dependency, an arbitrary value was defined as follows: high dependency, 2; intermediate dependency, 1; no dependency, 0. The degree of dependency of each patient was indicated in Table 1. (D) BCL2 dependency was compared between CCND1+ and CCND1− patients in the whole cohort (n = 47). (E) MCL1 dependency was analyzed in the CCND1, MMSET, and Others subgroups at diagnosis vs relapse. Fisher’s exact test was used for statistical analysis. (F) Percentage of no dependent patient samples was analyzed in translocated (CCND1/3, MAF, MMSET) and Others subgroups at diagnosis vs relapse. Fisher’s exact test was used for statistical analysis.
Dependence of primary MM samples on antiapoptotic BCL2 molecules, according to molecular subgroups. (A) Analyses of BCL2, MCL1, or BCLXL dependencies of 60 patients with MM at diagnosis and relapse. Patients’ dependencies to BCL2, MCL1, or BCLXL were defined by cell death response to the respective BH3 mimetic in primary myeloma cells, as indicated in Figure 1B and Table 1. Nodep includes patient samples that are insensitive to each 1 of the 3 BH3 mimetics (Table 1). Fisher’s exact test was used for statistical analysis. (B) High and intermediate patients’ dependencies to MCL1 were analyzed at diagnostic versus relapse. Fisher’s exact test was used for statistical analysis. (C) Analyses of individual dependencies of 60 patients with MM at diagnosis and relapse. For each dependency, an arbitrary value was defined as follows: high dependency, 2; intermediate dependency, 1; no dependency, 0. The degree of dependency of each patient was indicated in Table 1. (D) BCL2 dependency was compared between CCND1+ and CCND1− patients in the whole cohort (n = 47). (E) MCL1 dependency was analyzed in the CCND1, MMSET, and Others subgroups at diagnosis vs relapse. Fisher’s exact test was used for statistical analysis. (F) Percentage of no dependent patient samples was analyzed in translocated (CCND1/3, MAF, MMSET) and Others subgroups at diagnosis vs relapse. Fisher’s exact test was used for statistical analysis.
Of note, BCL2 dependency was significantly higher in CCND1 subgroup (83%) compared with all other subgroups (21%; P = .0001; Figure 2D). MCL1 dependency increased at relapse both in the CCND1 group and the Others group, but this increase was only significant in the latter group of patients (P = .03; Figure 2E). Furthermore, patients nondependent on any single antiapoptotic protein at diagnosis were mainly found in the group of patients who did not harbor a recurrent translocation (83%; P = .008; Figure 2F). Samples nondependent on any single antiapoptotic member decreased from 83% at diagnosis to 22% at relapse (P = .04; Figure 2F). These results indicate that nondependent primary cells were mainly found at diagnosis in the subgroup of patients not harboring a recurrent translocation. Finally, our findings highlight the predominance of MCL1 dependence at relapse either as an exclusive MCL1 dependence or as codependencies with BCL2 and/or BCLXL.
Analysis of the correlation between the expression of BCL2 family members and cell dependencies in patient samples
Among the 60 MM samples analyzed for cell dependencies, CD138+ MM cells from 41 patients have been purified and the expression of 3 main antiapoptotic genes (BCL2, BCLXL, and MCL1) was analyzed by quantitative polymerase chain reaction (supplemental Table 2). Because BCLXL and MCL1 have been shown to play a role in venetoclax resistance, we analyzed the correlation of venetoclax sensitivity (PC1 values; supplemental Figure 4A) with the ratio of BCL2/BCLXL, BCL2/MCL1, and BCL2/(BCLXL+MCL1) mRNA levels. Among them, we found that BCL2/BCLXL mRNA is the best marker of venetoclax sensitivity (r = 0.61; P = .0001; Figure 3A), indicating a major role of BCLXL in venetoclax resistance, as previously reported.13 To further define the involvement of antiapoptotic members in A1210477 response, we determined whether the ratio of MCL1/BCLXL, MCL1/BCL2, or MCL1/(BCLXL+BCL2) mRNA correlated with A1210477 sensitivity (PC1 values; supplemental Figure 4A), as we did not find any correlation between A1210477 sensitivity and the individual expression of MCL1 or BCLXL mRNA (supplemental Figure 4B). Although we found that only the MCL1/BCLXL mRNA ratio correlated with A1210477 sensitivity (r = 0.35; P = .02), this ratio could not be considered a biomarker of A1210477 response. Nevertheless, this finding indicates that a high BCLXL expression might be involved in A1210477 resistance. Altogether, these results suggest the contribution of BCLXL as a resistant factor for both venetoclax and A1210477 MCL1 BH3 mimetic.
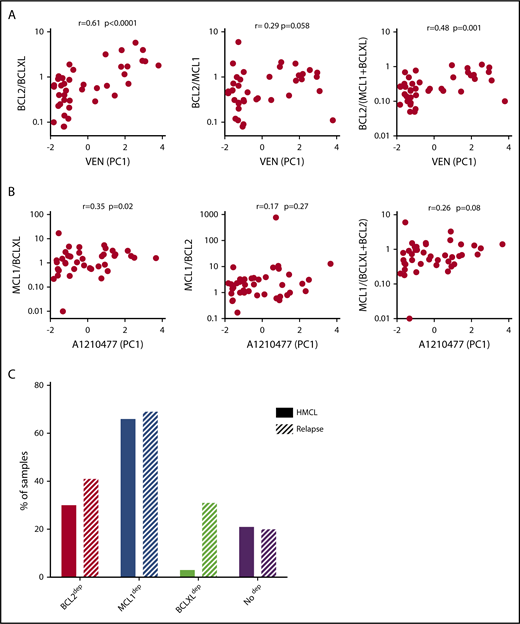
Analysis of BCL2 family members’ expression and cell dependencies in MM patient samples. (A) Analysis of BCL2/BCLXL, BCL2/MCL1, and BCL2/(MCL1+BCLXL) mRNA expression ratio in function of venetoclax sensitivity in patients with myeloma (n = 41). BCL2, BCLXL and MCL1 mRNA levels were defined by quantitative polymerase chain reaction, using Taq-Man probes (supplemental Table 2), and the different ratios were plotted against venetoclax sensitivity defined by the principal component (PC1) values (supplemental Figure 4A). The Spearman rank correlation is indicated. (B) Analysis of MCL1/BCLXL, MCL1/BCL2, and MCL1/BCLXL+BCL2 mRNA expression ratio in function of A1210477 sensitivity defined by the principal component (PC1) values in patients with myeloma (n = 41). Correlation was assessed by Spearman test. (C) Comparison of dependencies between HMCLs (n = 33) and patients at relapse (n = 39). Sensitivity to each BH3 mimetic of HMCLs is provided in Table 2.
Analysis of BCL2 family members’ expression and cell dependencies in MM patient samples. (A) Analysis of BCL2/BCLXL, BCL2/MCL1, and BCL2/(MCL1+BCLXL) mRNA expression ratio in function of venetoclax sensitivity in patients with myeloma (n = 41). BCL2, BCLXL and MCL1 mRNA levels were defined by quantitative polymerase chain reaction, using Taq-Man probes (supplemental Table 2), and the different ratios were plotted against venetoclax sensitivity defined by the principal component (PC1) values (supplemental Figure 4A). The Spearman rank correlation is indicated. (B) Analysis of MCL1/BCLXL, MCL1/BCL2, and MCL1/BCLXL+BCL2 mRNA expression ratio in function of A1210477 sensitivity defined by the principal component (PC1) values in patients with myeloma (n = 41). Correlation was assessed by Spearman test. (C) Comparison of dependencies between HMCLs (n = 33) and patients at relapse (n = 39). Sensitivity to each BH3 mimetic of HMCLs is provided in Table 2.
Dependencies of MM primary cells at relapse correlated with HMCL dependencies
Analysis of cell dependencies performed on 33 HMCLs showed that 30% of HMCLs were BCL2 dependent, 3% were BCLXL dependent, and the majority of HMCLs (66%) were MCL1 dependent (Table 2). Finally, 21% of HMCLs were nondependent on any single antiapoptotic member. In addition, only MM1S HMCL was efficiently killed by A1155463 (LD50 = 10 nM), showing that a minor subset of HMCL was highly BCLXL dependent (Table 2). Comparison of dependencies between primary myeloma cells at relapse and HMCLs showed a similitude (Figure 3C), indicating that dependencies of HMCLs mostly reflect the dependencies of primary myeloma cells observed at relapse, particularly highlighting the predominance of MCL1 dependency. Nevertheless, the BCXL dependency seems to be weakly represented in our myeloma cell line collection.
Sensitivity of HMCLs to the BH3 mimetics
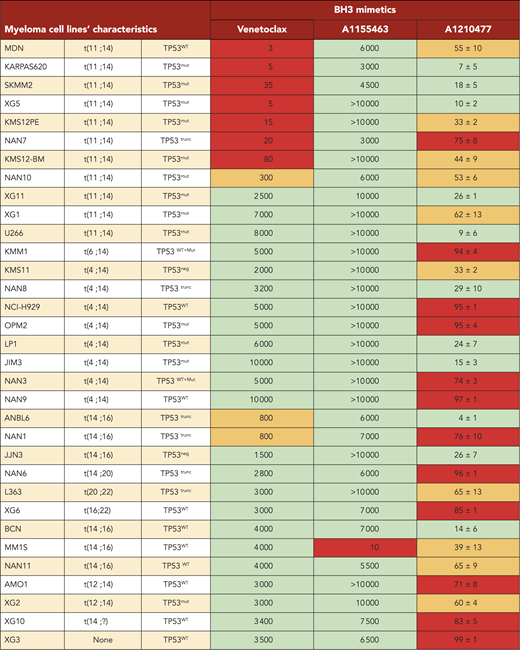 |
|
Viability was determined after 24 hours of treatment with increasing concentrations of venetoclax and A1155463 (0.001-10 μM) or A1210477 (5 μM). Cell death was assessed by flow cytometry after Annexin V staining. LD50 values for venetoclax (Ven) and A1155463 were calculated from 3 independent experiments. Apoptosis % induced by 5 μM A1210477 is indicated. Red, high (LD50 <300 nM for Ven and A115543, ≥65% apoptosis for 5 μM A1210477). Orange, intermediate (LD50 300 ≤1000 nM for Ven and A115543, 33-65% apoptosis for 5 μM A1210477). Green, not dependent (LD50 >1000 nM for Ven and A115543, <33% apoptosis for 5 μM A1210477).
Mechanism of action of the MCL1 BH3 mimetic in myeloma cells
Initially, we demonstrated that A1210477 induced apoptosis via the activation of the intrinsic apoptotic pathway, as shown by the release of cytochrome c (Figure 4A) and the activation of both caspase 9 and 3 (Figure 4B). To further understand the mechanism of action of the MCL1 BH3 mimetic, we compared the pharmacologic inhibition of MCL1 to BH3-profiling, using the NOXA/MS1 peptide specific for MCL1 (supplemental Table 3).15,23 Flow cytometry analysis demonstrated a robust and significant correlation (r = 0.79; P < .0001) between these 2 different approaches to define MCL1 dependency (Figure 4C). However, 2 cell lines had a strong mitochondrial response to NOXA/MS1 peptide, while displaying a weak A1210477 cell death induction, possibly indicating a defect in the apoptotic pathway downstream of the release of cytochrome c. The latter result underlines the interest of using BH3 mimetics that takes into account both the mitochondrial priming and the efficiency of the downstream apoptotic pathway. Because BAX and BAK are crucial for the efficient triggering of apoptosis, we studied the contribution of both effectors in A1210477 induced-cell death. BAK and BAX were transiently silenced in 2 MCL1-dependent cell lines (KMM1 and OPM2). BAK silencing significantly inhibited A1210477-induced cell death in both OPM2 (73% ± 6% inhibition) and KMM1 (44% ± 4% inhibition) cells, whereas the silencing of BAX did not protect against apoptosis induced by MCL1 BH3 mimetic (Figure 4D). We also analyzed the consequence of BIM silencing, and demonstrated that it did not have a major contribution to cell death induced by A1210477 (Figure 4D). We then analyzed the dynamics of MCL1 complexes induced by a short A1210477 treatment (1 and 3 hours). Although the treatment of A1210477 increased MCL1 protein levels, as already reported,8 it induced the dissociation of MCL1/BAK, MCL1/BIM, and MCL1/NOXA complexes (Figure 4E). The MCL1/BAK complexes were strongly decreased after 1 hour of A1210477 treatment, and totally disrupted after 3 hours of treatment (Figure 4E). Interestingly, complete inhibition of the MCL1/BAK interaction was observed in purified primary myeloma cells from patient #27 (Figure 4F).
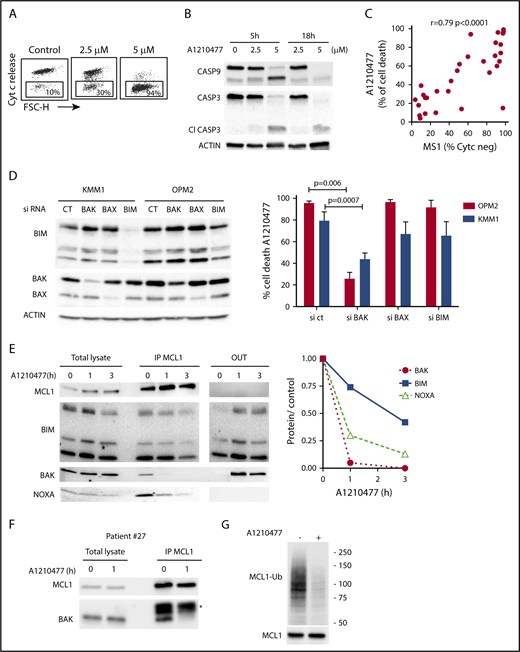
Mechanism of action of A1210477 in sensitive myeloma cells. (A) Cytochrome c release and (B) immunoblots of caspase 3 and 9 activation in OPM2 cell line under A1210477 treatment. Results are representative of at least 2 independent experiments. (C) Correlation between A1210477 sensitivity and BH3 profiling. BH3 profiling of HMCLs was performed using MS1 peptide (10 μM), and loss of cytochrome c was analyzed by flow cytometry, as previously described.20 Values of cytochrome c negative cells corresponding to BH3 profiling are indicated in supplemental Table 3. Sensitivity to A1210477 (5 μM) was plotted vs cytochrome c negative cells. The Spearman rank correlation is indicated. (D) A1210477-induced cell death is impaired by BAK silencing, but neither BAX nor BIM silencing. OPM2 and KMM1 were transfected with the different siRNA, protein expression was determined 48 hours after transfection, and cells were treated with A1210477 for 24 hours before assessing cell death by Annexin V staining. The induction of apoptosis was compared with the nontreated controls. Results represent the mean of 4 independent experiments. Statistical analysis was performed by Kruskal-Wallis test. (E) A1210477 disrupts the complexes of MCL1 with its proapoptotic counterparts. Immunoprecipitation of MCL1 was performed after short A1210477 treatment (2 μM) of OPM2 cells, followed by western blotting of indicated proteins. Quantification of proteins bound to MCL1 was performed for each condition relative to endogenous complexes without treatment. Quantification of bound proteins was performed using ImageJ software. (F) Myeloma cells from patient #27 were treated with A1210477 (2 μM) for 1 hour. Lysates were obtained and MCL1 immunoprecipitates were analyzed by western blot, as in E. *Ab light chain (G) A1210477 binding to MCL1 impaired MCL1 ubiquitination. OPM2 cell line was preincubated during 3 hours with MG-132 (1 μM). Then, A1210477 (1.5 μM) was added for 30 minutes. Cell lysates were used for the detection of ubiquitinylated MCL1 captured by TUBEs, followed by western blotting analysis.
Mechanism of action of A1210477 in sensitive myeloma cells. (A) Cytochrome c release and (B) immunoblots of caspase 3 and 9 activation in OPM2 cell line under A1210477 treatment. Results are representative of at least 2 independent experiments. (C) Correlation between A1210477 sensitivity and BH3 profiling. BH3 profiling of HMCLs was performed using MS1 peptide (10 μM), and loss of cytochrome c was analyzed by flow cytometry, as previously described.20 Values of cytochrome c negative cells corresponding to BH3 profiling are indicated in supplemental Table 3. Sensitivity to A1210477 (5 μM) was plotted vs cytochrome c negative cells. The Spearman rank correlation is indicated. (D) A1210477-induced cell death is impaired by BAK silencing, but neither BAX nor BIM silencing. OPM2 and KMM1 were transfected with the different siRNA, protein expression was determined 48 hours after transfection, and cells were treated with A1210477 for 24 hours before assessing cell death by Annexin V staining. The induction of apoptosis was compared with the nontreated controls. Results represent the mean of 4 independent experiments. Statistical analysis was performed by Kruskal-Wallis test. (E) A1210477 disrupts the complexes of MCL1 with its proapoptotic counterparts. Immunoprecipitation of MCL1 was performed after short A1210477 treatment (2 μM) of OPM2 cells, followed by western blotting of indicated proteins. Quantification of proteins bound to MCL1 was performed for each condition relative to endogenous complexes without treatment. Quantification of bound proteins was performed using ImageJ software. (F) Myeloma cells from patient #27 were treated with A1210477 (2 μM) for 1 hour. Lysates were obtained and MCL1 immunoprecipitates were analyzed by western blot, as in E. *Ab light chain (G) A1210477 binding to MCL1 impaired MCL1 ubiquitination. OPM2 cell line was preincubated during 3 hours with MG-132 (1 μM). Then, A1210477 (1.5 μM) was added for 30 minutes. Cell lysates were used for the detection of ubiquitinylated MCL1 captured by TUBEs, followed by western blotting analysis.
Although the MCL1/NOXA complexes were mostly disrupted at 3 hours, 40% of BIM still remained bound to MCL1 (Figure 4E), likely reflecting the higher affinity of BIM for MCL1.24 Because we observed that a short A1210477 treatment (1 hour) increased MCL1 protein levels, we analyze the ubiquitination status of MCL1. We demonstrated that a very short A1210477 treatment (30 min) induced a robust and rapid decrease of MCL1 ubiquitination, indicating that the accumulation of MCL1 under A1210477 treatment was reflected in an impaired MCL1 proteasome degradation (Figure 4G). This result is in agreement with previous data showing that A1210477 binding to MCL1 promotes a conformational switch in MCL1, leading to the inhibition of ubiquitination.25 Altogether, these results demonstrated that A1210477 induced apoptosis by dissociating endogenous MCL1/proapoptotic complexes while excluding a mechanism-mediated MCL1 degradation.
Contribution of BCLXL in the resistance to the MCL1 BH3 mimetic
Because it has been suggested that a high expression of BCLXL impaired A1210477-induced cell death,9 we assessed whether BCLXL was implicated in resistance to A1210477 in MM cells. We transiently silenced this antiapoptotic member in the BCLXL-dependent MM1S HMCL, as well as in the nondependent LP1 and U266 HMCLs. As observed in Figure 5A, the efficient BCLXL knockdown significantly sensitized the 3 HMCLs to the MCL1 BH3 mimetic; the strongest sensitization was found in MM1S BCLXL-dependent HMCL. To further study the contribution of BCLXL in A1210477 response, we tested the combination of low doses of both A1210477 and A1155463 in LP1 and U266 HMCLs. Interestingly, whereas these cell lines were insensitive to either inhibitor alone, the combined low dose of both drugs triggered apoptosis, showing that the combination was highly synergistic and that the pharmacologic inhibition of BCLXL could overcome A12104777 resistance (Figure 5B). To go deeper into BCLXL-induced resistance, we performed endogenous sequential immunoprecipitations after short-term exposure of the resistant MM1S and U266 HMCLs to A1210477. Analyses of MCL1 immunoprecipitations demonstrated, as in sensitive myeloma cells, that the endogenous interaction of BAK with MCL1 was completely dissociated after 1 hour of A1210477 treatment in both MM1S and U266 HMCLs (Figure 6A-B), whereas the interaction of BIM with MCL1 was not fully inhibited, even after 3 hours of treatment (Figure 6A-B). We next performed BCLXL immunoprecipitations, using the MCL1 depleted lysates. Interestingly, we found that BCLXL was able to recapture the fraction of BAK released from MCL1 under A1210477 treatment as early as 1 hour after treatment, as shown by the increased BAK bound to BCLXL in both MM1S and U266 HMCLs (Figure 6C-D). In addition, after BAK's recapture, BCLXL was also able to recapture BIM released from MCL1 (Figure 6C-D). These results suggest that BCLXL may act as a sink to bind freed proapoptotic proteins from MCL1 and limit MM cell death triggered by the specific targeting of MCL1.
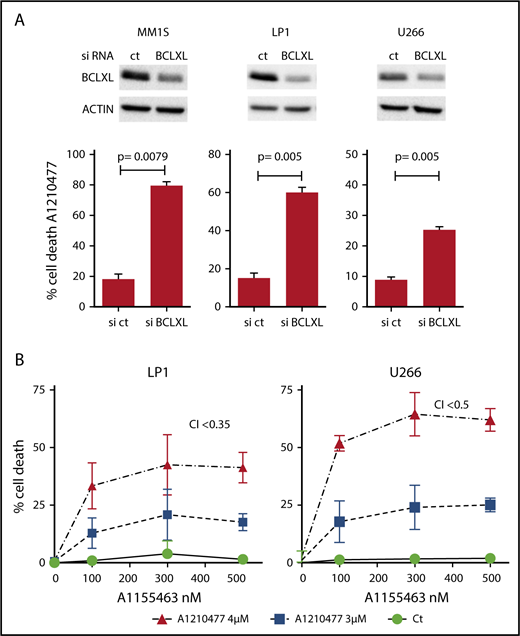
BCLXL is the major factor that limits MM cell death to the pharmacological inhibition of MCL1. (A) After transfection with scramble or BCLXL-specific siRNA, MM1S, LP1, and U266 HMCLs were treated with A1210477 (5 μM) for 24 hours, cell death was assessed by Annexin V staining and the induction of apoptosis was compared with the non-treated controls. Results represent the mean ± SD of 5 independent experiments. Statistical analysis was performed using Student t-Test. (B) Combination of A1210477 and A1155463 overcomes A1210477 resistance in LP1 and U266 HMCLs. LP1 and U266 HMCLs were treated with combination of low doses of both A1210477 (3 and 4 μM) and A1155463 (100, 300 and 500 nM). All data points represent the mean of triplicate experiments ± SD. Combination index (CI) was calculated using CalcuSyn software.
BCLXL is the major factor that limits MM cell death to the pharmacological inhibition of MCL1. (A) After transfection with scramble or BCLXL-specific siRNA, MM1S, LP1, and U266 HMCLs were treated with A1210477 (5 μM) for 24 hours, cell death was assessed by Annexin V staining and the induction of apoptosis was compared with the non-treated controls. Results represent the mean ± SD of 5 independent experiments. Statistical analysis was performed using Student t-Test. (B) Combination of A1210477 and A1155463 overcomes A1210477 resistance in LP1 and U266 HMCLs. LP1 and U266 HMCLs were treated with combination of low doses of both A1210477 (3 and 4 μM) and A1155463 (100, 300 and 500 nM). All data points represent the mean of triplicate experiments ± SD. Combination index (CI) was calculated using CalcuSyn software.
![Figure 6. BCLXL acts as a sink to capture freed proapoptotic proteins from MCL1 under A1210477 treatment in MCL1 nondependent cell lines. (A) MM1S and (B) U266 A1210477-resistant HMCLs were treated with A1210477 (2 μM) for the indicated times. Immunoprecipitation reactions of MCL1 were performed followed by western blotting analysis of BAK and BIM. # A minor BIM isoform other than EL, L or S associated with MCL1 in MM1S. Quantification of bound BAK and BIM proteins to MCL1 was done using ImageJ software. ][An empty space has been removed from the original image. (C-D) Free-MCL1 lysates of MM1S and U266 cell lines were then subjected to BCLXL immunoprecipitation reactions followed by western blotting analysis of BAK and BIM. *Ab light chain. Quantification of bound BAK and BIM proteins to BCLXL was done as in A and B.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/25/10.1182_blood-2018-03-836718/5/m_blood836718f6.png?Expires=1769220865&Signature=GnzPb4nTWECoe-8Y0L6-iBO27kPFssHVMfkt1Mn3QlRIYfxEKBgxlJ2obTnhnDBGuWzAH4bXar9wT0ThLOjXtTaSbTqKwQQndDQ1BhRE2rL2iHBGGl2xowA-m7OOiQ~z-S8iIZnqYZHDMxQg~tQReT3XQZzX1w~I6xE5grsxImLdNLWbTYWk5jPMfA1GmezUKmni~1-I75sC~1eFX5HRl9FhnhwE6bKMaG5QjWdwWV70luIEWn1GwluvgpsTeRji6u1bDepHqCjI6GLh6dbkeAFjfLaBxMo~OGIrZFTQ8bUpYYOv8drfB8Wy5Gp0xctukqPvY66hQCPLPf0Em-~5MQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
BCLXL acts as a sink to capture freed proapoptotic proteins from MCL1 under A1210477 treatment in MCL1 nondependent cell lines. (A) MM1S and (B) U266 A1210477-resistant HMCLs were treated with A1210477 (2 μM) for the indicated times. Immunoprecipitation reactions of MCL1 were performed followed by western blotting analysis of BAK and BIM. # A minor BIM isoform other than EL, L or S associated with MCL1 in MM1S. Quantification of bound BAK and BIM proteins to MCL1 was done using ImageJ software. ][An empty space has been removed from the original image. (C-D) Free-MCL1 lysates of MM1S and U266 cell lines were then subjected to BCLXL immunoprecipitation reactions followed by western blotting analysis of BAK and BIM. *Ab light chain. Quantification of bound BAK and BIM proteins to BCLXL was done as in A and B.
BCLXL acts as a sink to capture freed proapoptotic proteins from MCL1 under A1210477 treatment in MCL1 nondependent cell lines. (A) MM1S and (B) U266 A1210477-resistant HMCLs were treated with A1210477 (2 μM) for the indicated times. Immunoprecipitation reactions of MCL1 were performed followed by western blotting analysis of BAK and BIM. # A minor BIM isoform other than EL, L or S associated with MCL1 in MM1S. Quantification of bound BAK and BIM proteins to MCL1 was done using ImageJ software. ][An empty space has been removed from the original image. (C-D) Free-MCL1 lysates of MM1S and U266 cell lines were then subjected to BCLXL immunoprecipitation reactions followed by western blotting analysis of BAK and BIM. *Ab light chain. Quantification of bound BAK and BIM proteins to BCLXL was done as in A and B.
Discussion
The BH3 mimetics ex vivo assay of primary myeloma cells, analyzed by an unbiased approach of cell death clustering, allowed the identification of subgroups with specific dependencies on antiapoptotic BCL2 proteins. We found a significant increase in MCL1 dependency from diagnosis to relapse, mostly as a result of the increase in intermediate-dependent MCL1 samples. Further, we identified a group of patient samples not sensitive to any of the 3 BH3 mimetics. Unexpectedly, a large proportion of those samples was found in the diagnosis group. We showed that BCLXL dependency was minor and rarely alone, but often accompanied with a codependency either on BCL2 and/or MCL1. We confirmed previous findings showing that BCL2 dependence was mostly found in the CCND1 subgroup of patients, but also extended to other subgroups.
The striking increase in MCL1 dependency or codependency at relapse, mostly found in the group of patients lacking recurrent translocations, but also in the CCND1 group, suggests a plasticity of the cellular dependency toward MCL1 in these specific groups. It appears, therefore, that previous treatments or clonal selection during the course of the disease could favor MCL1 dependency. The predominance of MCL1 dependence also found in HMCLs, as previously reported,26 is probably because all HMCLs were generated from relapsed patients, mainly with extramedullary disease. Therefore, extrapolation of results obtained on HMCLs for preclinical purpose must be done with caution.
BCL2 dependency was mainly found in the CCND1 molecular subgroup, either at diagnosis or at relapse, and was characterized by a high BCL2/BCLXL mRNA expression, as already reported.13,14 Because a significant proportion of CCND1 patients with MM at diagnosis are only sensitive to venetoclax, therapeutic intervention targeting BCL2 could be proposed from the early phase of the disease. It should be noted that the incidence of CCND1 patients is slightly higher (37%) in our cohort than the reported incidence for MM.27 At relapse, more than 40% of patients showed different codependencies, suggesting that these patients could be potentially targeted by either venetoclax or MCL1 BH3 mimetics. Because targeting BCLXL with a BH3 mimetic remains a problem in clinic because of the induction of thrombocytopenia,28 it is interesting to note that BCLXL dependency of patients with MM was rarely exclusive, and MCL1 and BCL2 mimetics could be potentially used in these patients. Of note, the identification of a group of patient samples not sensitive to any of the 3 BH3 mimetics confirmed a similar observation reported by Touzeau et al.15 Interestingly, a large proportion of these samples was found at diagnosis and decreased at relapse. In addition, most of them did not harbor recurrent translocations. These findings suggest that nondependent patients could acquire dependencies on antiapoptotic proteins during the progression of the disease. They also show the extraordinary ability of tumor adaptation to conventional therapy, highlighting the interest of targeting antiapoptotic proteins, and particularly MCL1, at relapse stage.
Our mechanistic studies showed that MCL1 BH3 mimetic killed myeloma cells in a BAK-dependent manner and led to the complete disruption of BAK/MCL1 and NOXA/MCL1 complexes. We also demonstrated that A1210477 treatment decreased MCL1 ubiquitination in agreement with the results of in vitro ubiquitination assays already reported by Song et al.25 The preferential role of BAK vs BAX in apoptosis induced by MCL1 BH3 mimetic was already reported in other models9,29 and consistent with our previous findings demonstrating the privileged role of BAK in MM cell death induced on endoplasmic reticulum stress.30 The exclusive BAK implication is in contrast with data reported in Hela cells showing that the pharmacologic inhibition of MCL1 killed cancer cells in a BAX- and BAK-dependent manner.10 This result should be carefully analyzed because MCL1 was overexpressed in Hela cells; thus, the analysis of cells under endogenous conditions could eventually warrant a more reliable conclusion. Of interest, we unraveled the mode of innate resistance to A1210477-induced cell death and demonstrated the major implication of BCLXL in this process. Indeed, the dual pharmacological inhibition of BCLXL and MCL1 was found to be highly synergistic, reinforcing the role of BCLXL in MCL1 BH3-mimetic resistance. Furthermore, the dissociation pattern of MCL1/proapoptotic proteins in resistant MM cell lines was similar to that observed in myeloma-sensitive cells. However, released proapoptotic proteins were recaptured by BCLXL, explaining the role of BCLXL in the innate resistance to A1210477. An analogous mechanism of redistribution of proapoptotic proteins from both BCL2 and BCLXL to MCL1 was observed on ABT-737 treatment.31
Because the knockout of MCL1 in a murine model causes hepatic, hematologic, and cardiac toxicities, including rapid development of heart failure,32,33 the question of MCL1 BH3 mimetics tolerability remains crucial. The fact that MCL1 BH3 mimetics affect only the ability of MCL1 to sequester proapoptotic proteins and not the other additional roles of MCL1, such as its implication in the mitochondrial respiration,34 may in part explain its tolerance in animal models.10,29 Furthermore, the episodic inhibition of MCL1 is also enforced by the stabilization of MCL1 under the binding of A1210477 or S63845 BH3 mimetics. Accordingly, we could hypothesize that the stabilization of MCL1 under the binding of BH3 mimetics might also be beneficial for its clinical application. However, we cannot rule out a detrimental effect resulting from the stabilization of MCL1, impairing its efficacy. Further preclinical studies are necessary to better understand the mechanism of MCL1 BH3 mimetics that warrant its safety in therapeutic application.
In conclusion, our study highlights the ex vivo testing of primary myeloma cell dependencies, using the BH3 toolkit as a potential guide for the respective and tailored use of venetoclax and MCL1 BH3 mimetics in myeloma at diagnosis and/or relapse. Although this functional assay requires viable cells, it can be broadly applicable, as it is conducted as a simple cell viability assay analyzed by flow cytometry. Finally, the analysis of a larger cohort of patients should be mandatory to further tailor the use of the appropriate BH3 mimetic, according to the heterogeneity of the disease represented by the common cytogenetic subtypes, as well as the secondary cytogenetic abnormalities associated with adverse prognosis.35
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the flow cytometry core facility (Cytocell, SFR Bonamy), C. Godon, O. Theisen, and M. Eveillard for expertise in fluorescence in situ hybridization analysis, and M. Loirat (CHD of Saint-Nazaire) and N. Morineau (CHD of La Roche/Yon) for providing myeloma samples. The authors also thank Kathryn Jacobs (CRCINA) for proofreading the manuscript.
This study was supported by the SIRIC ILIAD, INCa-DGOS-Inserm_12558, Ligue Contre le Cancer Grand-Ouest, FFRMG, and by Actions Cancer 44. B.T. was supported by INSERM (poste d’accueil) and Fondation ARC.
Authorship
Contribution: P.G.-B., S.M., B.T., J.B., and A.B. performed experiments and analyzed the results; C.T analyzed the results; S.L.G., C.T., and P.M. provided primary myeloma samples and reviewed the paper; M.S.R. provided tandem ubiquitin binding entities and analyzed ubiquitination results; and P.G.-B., B.T., C.P.-D., and M.A. designed the research and wrote the paper.
Conflict-of-interest disclosure: P.M. serves on advisory boards for Celgene, Janssen, Novartis, and AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Martine Amiot, CRCINA, INSERM UMR1232, CNRS ERL6001, 8, quai Moncousu, BP 70721, 44007, Nantes, France; e-mail: martine.amiot@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal