

Abstract
Background: Point mutations of RUNX1 gene are frequently seen in myeloid malignancies such as myelodysplasia (MDS) and acute myeloid leukemia (AML), and are associated with unfavorable clinical outcomes. AML patients bearing RUNX1 mutations have a significantly lower complete remission (CR) rate compared with patients with wild-type RUNX1 (30% vs. 73%), whereas in contrast, the CR rate of ALL patients, in which RUNX1 is rarely mutated, are at 92%. It remains unclear whether RUNX1 mutations found in AML patients are causal to AML maintenance, and if RUNX1 mutations serve as a valid therapeutic target in the treatment of such therapy-resistant AML.
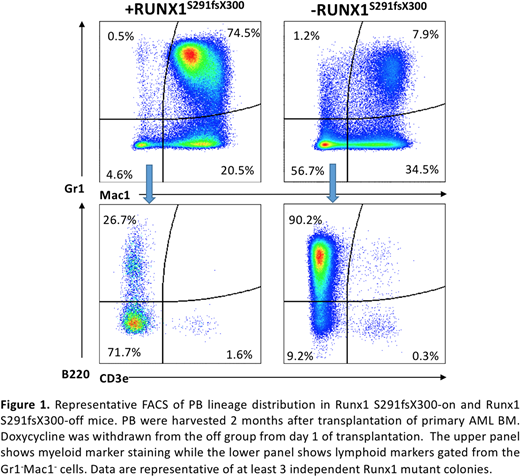
Experimental Approach: The RUNX1 mutations are found frequently associated with MLL-partial tandem duplication (MLL-PTD) in AML. We have developed two complementary mouse models that express a MLL-PTDknock-in mutation together with a RUNX1 S291fsX300 mutation found in AML patients: in the first model RUNX1 S291fsX300 was introduced into the bone marrow cells of the MLL-PTD knock-in mice by retrovirus transduction, while in the second model the tetracycline-inducible RUNX1 S291fsX300 mutation was knock-in at the Collagen a1 locus and the mice was crossed to MLL-PTD knock-in. In the second model, the mutant RUNX1 protein is only expressed when the mice were fed with doxycycline containing food and the RUNX1 mutant is absent or "corrected" upon doxycycline withdrawn. BM cells of these mice, as well as WT and MLL-PTD controls, were transplanted to syngeneic mice and the recipients were tracked for disease development and progression at bi-monthly intervals. The doxycycline induction or withdraw was carried out after secondary transplant, and the expression of RUNX1 S291fsX300 was monitored by a built-in GFP reporter and verified by RT-PCR.Under tetracycline induction, the RUNX1 S291fsX300 and MLL-PTD double mutation bearing mice developed a spontaneously AML and had a survival time 6-10 months after transplantation, and they showed symptoms of MDS/AML or AML, including thrombocytopenia, anemia, leukocytosis, splenomegaly, abnormal BM and spleen cell morphologies. We transplanted the AML mouse bone marrow to secondary recipients, and observed them with or without continuing doxycycline induction. By using the O-propargyl-puromycin to track the newly synthesized proteins, we found that the RUNX1 mut-on and RUNX1 mut-off mice showed comparable protein synthesis rate at full-blown leukemia stage, indicating that the RUNX1 mutation does not play a restrictive role to protein synthesis in transformed leukemia cells. The c-Kit+ AML cells cultured from the RUNX1 mut-off mice showed reduced viability and significantly reduced CFU-C activities, which are accompanied by a lineage switch in a decrease in expression of Mac1 and an increase in expression of B220. In vivo, the RUNX1 mut-off mice showed a slightly prolonged survival compared with RUNX1 mut-on group (182 vs. 126 days, p=0.0036). An analysis of the lineage distribution in their peripheral blood and bone marrow revealed that the RUNX1 mut-off AML mice underwent a dramatic lineage switch from myelocytes to lymphocytes (Fig. 1): while the Mac1+Gr1+population remained high in the RUNX1 mut-on AML mice, turning off RUNX1 mut expression caused an increased B cell population in expense of the Mac1+Gr1+population. Pathological analysis also showed a drastic decrease of the myeloid marker MPO and increased lymphoid marker B220 in the RUNX1mu-off mice. Consistently, a RT-PCR test of the BM c-Kit+ cells further showed that upon turning off the RUNX1mut, critical B cell regulatory genes including TCF3, EBF1, PAX5, CD79A were significantly up regulated whereas myeloid regulatory genes such as PU.1 were downregulated. In addition, RUNX1 S291fsX300 expression in the AML cells confers a resistance to chemotherapy (Ara-C) treatment compared with that of WT RUNX1 expression, indicating that the RUNX1 mutation is responsible for the AML resistance to therapy.
Conclusion:Although RUNX1 mutations found in AML patient may not be essential for the hyperproliferative and growth phenotype of the AML, they play a causal role in maintaining myeloid lineage restriction, expansion and chemo-resistance. Correction or removal of the RUNX1 mutation in AML cells leads to a disease switch from AML to B-ALL, effectively changing the difficult-to-treat drug-resistant AML to another disease with more favorable CR (B-ALL).
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal