

Abstract
Myelodysplastic syndromes (MDS) are clonal diseases defined by clinical, morphologic, and genetic features often shared by related myeloid disorders. The diagnostic boundaries between these diseases can be arbitrary and not necessarily reflective of underlying disease biology or outcomes. In practice, measures that distinguish MDS from related disorders may be difficult to quantify and can vary as disease progression occurs. Patients may harbor findings that are not consistent with a single diagnostic category. Several overlap disorders have been formally described, such as the myelodysplastic/myeloproliferative neoplasms (MDS/MPNs). These disorders are characterized by hematopoietic dysplasia with increased proliferation of monocytes, neutrophils, or platelets. They may have mutational profiles that distinguish them from the disorders they resemble and reflect important differences in pathophysiology. MDS also shares diagnostic borders with other diseases. For example, aplastic anemia and hypoplastic MDS can be difficult to distinguish in patients with pancytopenia and bone marrow hypocellularity. Genetic features may help in this regard, because they can identify differences in prognosis and risk of progression. The boundary between MDS and secondary acute myeloid leukemia (sAML) is arbitrarily defined and has been redefined over the years. Genetic studies have demonstrated that sAML clones can precede clinical progression from MDS by many months, suggesting that MDS with excess blasts could be viewed as an overlap between a dysplastic bone marrow failure syndrome and an oligoblastic leukemia. This review will describe the diagnostic boundaries between MDS, MDS/MPNs, sAML, clonal hematopoiesis of indeterminate potential, clonal cytopenia of undetermined significance, and aplastic anemia and how genetic approaches may help to better define them.
Introduction
Myelodysplastic syndromes (MDS) are clonal hematopoietic disorders that typically present with features indicative of bone marrow failure, including inefficient hematopoiesis, morphologic dysplasia, and cytopenias of the peripheral blood. This clinical phenotype is nonspecific and can be a consequence of a variety of benign or malignant conditions. MDS and its mimics show an increased incidence with age, often making it challenging to arrive at the appropriate diagnosis. This is exacerbated by the fact that the apparently well-defined diagnostic boundaries between MDS and related conditions can, in practice, be much more vague and difficult to characterize. This can occur at initial presentation, when a patient with MDS-like features might also have evidence of hypoplasia or a myeloproliferative neoplasm (MPN), and over time, because MDS can evolve into another diagnosis, such as a secondary acute myeloid leukemia (sAML). Recent advances in our understanding about the genetics of MDS and its diagnostic neighbors may help to sharpen their boundaries or, ultimately, redefine them altogether.
Correctly diagnosing patients with MDS overlap syndromes can have important clinical implications. Often, the prognosis associated with an overlap syndrome is distinct from the individual disorders that they resemble. This is driven, in part, by differences in their genetic profiles and pathobiology. Consequently, overlap disorders may be amenable to different therapeutic options and can harbor unique molecular vulnerabilities. Although genetics can aid in the diagnosis of overlap disorders, somatic mutations rarely define them independent of the clinical context in which they are found. Other factors, including patient characteristics, epigenetic alterations, and microenvironmental interactions, such as inflammation and adaptive immune responses, help to shape the disease phenotype. Together, these characteristics can help to establish an accurate diagnosis in cases with overlapping features.
This review will focus on those diagnostic categories that have features of MDS combined with elements of other disorders in the context of our greater understanding about their underlying molecular genetics. This includes the individual MDS/MPN overlap disorders recognized in the World Health Organization (WHO) classification of myeloid neoplasms. We will also examine the diagnostic boundaries between MDS and two related conditions, aplastic anemia (AA) and sAML, disorders that can lead to, or arise from, MDS respectively. Finally, we explore the diagnostic boundaries between lower-risk MDS, clonal hematopoiesis of indeterminate potential (CHIP), and idiopathic cytopenias of undetermined significance (ICUS), a substantial fraction of which harbor somatic mutations typical of MDS.
The MDS/MPN overlap disorders
MDS/MPN overlap disorders are considered distinct from MDS and MPN, according to the WHO classification scheme. They include diagnoses with very different clinical manifestations, underlying genetics, and overall prognosis. Their shared features can include cellular dysplasia or cytopenias, in addition to an elevation in ≥1 blood cell count. At the molecular level, MDS/MPN disorders are more likely to carry gene mutations associated with the activation of growth factor signaling pathways in conjunction with mutations in epigenetic regulators or splicing factors associated with morphologic dysplasia.
Chronic myelomonocytic leukemia (CMML) is the most common of the MDS/MPN overlap diseases, even though its prevalence is estimated to be only ∼10% of that for MDS. CMML is defined by the presence of monocytosis in addition to ≥1 cytopenia (typically anemia) and bone marrow findings that typically meet MDS diagnostic criteria. The monocytosis in CMML has to be relative (≥10% of white blood cells) and absolute (≥1 × 109/L) and must persist for ≥3 months.1,2 Criteria indicative of other myeloid neoplasms and alternative causes of monocytosis should be absent. Historically, the French–American–British classification scheme considered CMML to be a subtype of MDS. The subsequent WHO classification divided CMML into a “proliferative type,” with a total white blood cell (WBC) count ≥13 × 109/L, and a “dysplastic type,” with a WBC count below this threshold, to reflect clinical and genetic distinctions between these 2 subtypes.3,4 In the most recent WHO classification, CMML is considered a separate entity from MDS and is classified into subtypes based on blood and bone marrow blast proportions and not on total WBC count (Table 1).
Clinical features associated with different MDS/MPN overlap conditions and disorders at the diagnostic boundary with MDS
| Median age (y) | Female/male ratio | Laboratory features | Physical features | Bone marrow features | |||
|---|---|---|---|---|---|---|---|
| Dysplasia | Cellularity | Other | |||||
| MDS | 70-71 | 1:1.5 | Anemia is the most common cytopenia seen, 25-40% have thrombocytopenia | Hepatosplenomegaly or extramedullary involvement not seen | Present | Hypercellular (10-20% hypocellular) | Dysplasia in ≥1 cell lineage; <20% blasts; 10% may have fibrosis |
| CMML | 65-75 | 1:1.5-3 | Absolute monocyte count ≥1.0 × 109/L, accounting for ≥10% of total WBCs for ≥3 mo | Splenomegaly present in up to half of patients; hepatomegaly and extramedullary involvement (skin, LNs) may be seen | Typically present but not required for diagnosis | Hypercellular | Dysplasia typical in ≥1 cell lineage, but may be absent; <20% blasts |
| MDS/MPN-RS-T | 72-73 | 1:1 | Anemia and platelet count ≥450 × 109/L | Thromboembolism may occur | Present | Hypercellular | ≥15% erythroid precursors with ring sideroblasts; megakaryocytic atypia; <5% blasts |
| aCML | 69-72 | 1:1.5 | WBCs >13 × 109/L, increased dysplastic neutrophils, no or minimal monocytosis and basophilia | Splenomegaly may be present | Present | Hypercellular | Dysplasia in ≥1 cell lineage; <20% blasts |
| JMML | 1-2 | 1:2-3 | Absolute monocyte count ≥1.0 × 109/L accounting for ≥10% of total WBCs for ≥3 mo | Splenomegaly common; monocytic and granulocytic infiltration of LNs, liver, skin, GI tract, and lungs also seen | Present | Hypercellular | <20% blasts |
| sAML | 70-73 | 1:1.5 | Variety of peripheral blood cytopenias may be seen, with or without leukocytosis | Hepatosplenomegaly may be present; infiltration of skin, gingiva, and CNS common in monocytic subtypes | Present | Hypercellular | ≥20% blasts; Auer rods |
| SAA | 50% <50 y old | 1:1 | Pancytopenia | Hepatosplenomegaly is not common; congenital anomalies may suggest an inherited marrow failure syndrome | Absent | Hypocellular | Profoundly hypocellular with all myeloid cell lineages diminished; marrow primarily composed of fat/stroma |
| CCUS | 65-75 | 1:1.5 | Anemia most common; other cytopenias can occur, often isolated | Hepatosplenomegaly or extramedullary involvement not seen | Absent or minimal | Normocellular or hypercellular | Does not meet MDS diagnostic criteria; fewer mutations in MDS genes but with comparable VAF |
| Median age (y) | Female/male ratio | Laboratory features | Physical features | Bone marrow features | |||
|---|---|---|---|---|---|---|---|
| Dysplasia | Cellularity | Other | |||||
| MDS | 70-71 | 1:1.5 | Anemia is the most common cytopenia seen, 25-40% have thrombocytopenia | Hepatosplenomegaly or extramedullary involvement not seen | Present | Hypercellular (10-20% hypocellular) | Dysplasia in ≥1 cell lineage; <20% blasts; 10% may have fibrosis |
| CMML | 65-75 | 1:1.5-3 | Absolute monocyte count ≥1.0 × 109/L, accounting for ≥10% of total WBCs for ≥3 mo | Splenomegaly present in up to half of patients; hepatomegaly and extramedullary involvement (skin, LNs) may be seen | Typically present but not required for diagnosis | Hypercellular | Dysplasia typical in ≥1 cell lineage, but may be absent; <20% blasts |
| MDS/MPN-RS-T | 72-73 | 1:1 | Anemia and platelet count ≥450 × 109/L | Thromboembolism may occur | Present | Hypercellular | ≥15% erythroid precursors with ring sideroblasts; megakaryocytic atypia; <5% blasts |
| aCML | 69-72 | 1:1.5 | WBCs >13 × 109/L, increased dysplastic neutrophils, no or minimal monocytosis and basophilia | Splenomegaly may be present | Present | Hypercellular | Dysplasia in ≥1 cell lineage; <20% blasts |
| JMML | 1-2 | 1:2-3 | Absolute monocyte count ≥1.0 × 109/L accounting for ≥10% of total WBCs for ≥3 mo | Splenomegaly common; monocytic and granulocytic infiltration of LNs, liver, skin, GI tract, and lungs also seen | Present | Hypercellular | <20% blasts |
| sAML | 70-73 | 1:1.5 | Variety of peripheral blood cytopenias may be seen, with or without leukocytosis | Hepatosplenomegaly may be present; infiltration of skin, gingiva, and CNS common in monocytic subtypes | Present | Hypercellular | ≥20% blasts; Auer rods |
| SAA | 50% <50 y old | 1:1 | Pancytopenia | Hepatosplenomegaly is not common; congenital anomalies may suggest an inherited marrow failure syndrome | Absent | Hypocellular | Profoundly hypocellular with all myeloid cell lineages diminished; marrow primarily composed of fat/stroma |
| CCUS | 65-75 | 1:1.5 | Anemia most common; other cytopenias can occur, often isolated | Hepatosplenomegaly or extramedullary involvement not seen | Absent or minimal | Normocellular or hypercellular | Does not meet MDS diagnostic criteria; fewer mutations in MDS genes but with comparable VAF |
CNS, central nervous system; GI, gastrointestinal; LN, lymph node.
Despite what seems like an arbitrary numerical distinction between MDS and CMML, there is evidence that the underlying pathobiology in these disorders is quite different. At the cellular level, patients with CMML have a high percentage of classical monocytes that are CD14+ and CD16−.5,6 These cells show a hypersensitivity to growth factor stimulation with granulocyte-macrophage colony-stimulating factor that is not observed in MDS. At the genetic level, CMML patients also have distinct mutational profiles (Figure 2). Somatic mutations in several genes, including TET2, ASXL1, SRSF2, EZH2, NRAS, KRAS, and CBL, are significantly more common in patients with CMML and, therefore, are more likely to co-occur.7-9 In fact, the triad of TET2, ASXL1, and SRSF2 mutations is highly specific for CMML.10-12 In contrast, mutations of SF3B1 and TP53 are observed less often than in MDS.
Clinically, patients with proliferative type CMML are enriched for RAS signaling pathway mutations and appear to have slightly greater disease-related risk than MDS patients with similar blast proportions.10,13 The original and revised International Prognostic Scoring Systems only included a small fraction of patients with dysplastic CMML (WBC <13 × 109/L), whereas proliferative CMML was excluded.14,15 This has led to the development of several CMML-specific prognostic tools.10,16-19 Where these models consider molecular abnormalities, mutations of ASXL1 are universally identified as independent adverse prognostic markers.
Therapeutic approaches for CMML aim to improve symptoms related to peripheral cytopenias or blood count proliferation. Similar to patients with MDS, hypomethylating agents may be considered for patients with CMML if poor prognostic factors or excess blasts are present. Response rates and benefit from treatment with hypomethylating agents appear to be comparable between patients with CMML and MDS.20,21 Surprisingly, responding CMML patients can revert to a normal monocyte profile with improved blood counts, without demonstrating changes in clonal burden.22 And unlike in MDS, DNA-methylation profiles predictive of hypomethylating agent response have been identified in CMML.23 Allogeneic hematopoietic stem cell transplantation (HSCT) remains the only curative treatment for CMML and should be considered in younger patients with higher-risk CMML, although the increased use of reduced-intensity conditioning and alternative donor sources has allowed increased implementation of HSCT in older patients. Expert opinion, including a recent international panel, suggests treatment before HSCT, particularly when marrow blasts are >10% or other higher-risk features are present.24,25
The next most common MDS/MPN overlap disorder is MDS/MPN with ring sideroblasts and thrombocytosis (MDS/MPN-RS-T). This entity has very little resemblance to CMML, despite being in the same diagnostic category. Patients with MDS/MPN-RS-T meet criteria for MDS with ring sideroblasts ≥15% and must have a persistently elevated platelet count (≥450 × 109/L). Classical hotspot mutations of SF3B1 are found in >80% of cases, resembling the rate of SF3B1 mutation observed in MDS-RS patients with single-lineage dysplasia (MDS-RS-SLD), and are often the likely founder mutation based on variant frequency and occurrence as the sole abnormality in some patients.26 MDS/MPN-RS-T patients also have a high rate of mutations in JAK2 (50-70%), CALR (10-20%), and MPL (2-5%), comparable to the mutational spectrum observed in essential thrombocythemia (ET).27,28 Mutations in several other genes may be present, including TET2, DNMT3A, ASXL1, and SETBP1, with the latter 2 being considered prognostically adverse.29 The prognosis of patients with MDS/MPN-RS-T lies between that of patients with MDS-RS-SLD and ET; the leukemic transformation rate per 100 years is similar in MDS/MPN-RS-T (1.8) and MDS-RS-SLD (2.4) and is higher in MDS/MPN-RS-T compared with ET (0.7).30 Rates of thrombosis are similar in MDS/MPN-RS-T and ET but are higher than in MDS-RS.30,31 In patients with anemia, treatment is usually supportive, with erythropoiesis-stimulating agents and transfusions following guidelines for lower-risk MDS. Low-dose aspirin may be prescribed for patients with JAK2 mutations, older age, or cardiovascular risk factors. Although del(5q) is not common in MDS/MPN-RS-T, case reports have described activity of lenalidomide, a drug that typically causes thrombocytopenia.32 Cytoreductive therapy is generally avoided because it can exacerbate anemia, but it may be implemented in the presence of multiple risk factors for thrombosis, vasomotor symptoms, or acquired von Willebrand syndrome.
Atypical chronic myeloid leukemia (aCML) is another WHO-recognized MDS/MPN overlap syndrome that is characterized by leukocytosis.33 As its name suggests, it is distinct from classical chronic myeloid leukemia (CML) driven by the BCR-ABL1 fusion gene. Specifically, aCML requires some degree of dysgranulopoiesis in the blood and bone marrow, minimal or no absolute basophilia (common in CML), minimal or no absolute monocytosis (common in CMML), and no gene rearrangements associated with other neoplasms (eg, BCR-ABL1, PDGFRA, PDGFRB, FGFR1, or PCM1-JAK2). Mutations typical of BCR-ABL1− MPN, like those in JAK2, CALR, and MPL, make a diagnosis of aCML less likely. The same is true for mutations of CSF3R, which are found in <10% of patients with aCML compared with 80% to 90% of patients with chronic neutrophilic leukemia, a clinically similar disorder that also presents with leukocytosis but no dysgranulopoiesis.33-35 No single molecular abnormality specific for aCML has been described, although SETBP1 mutations occur more frequently in aCML (25%) compared with CMML (6-15%) and JMML (3%).36 Recurrent mutations in several other CMML-like genes have also been detected in patients with aCML (Figure 2).37 In general, aCML patients tend to have a more aggressive disease course compared with patients with MDS/MPN, unclassifiable.33 Although there is no consensus on the role of HSCT, long-term remissions have been reported with this strategy.38 Other commonly used treatments include hypomethylating agent therapy and cytoreduction with hydroxyurea. The investigational use of JAK inhibitors has also been implemented in aCML and chronic neutrophilic leukemia based on the knowledge that some CSF3R mutations, most commonly CSF3RT618I, may activate the JAK/STAT pathway.39
Juvenile myelomonocytic leukemia (JMML) is an uncommon MDS/MPN overlap syndrome that occurs in early childhood, with a median age of 2 years. Clinical outcomes vary in JMML, with a minority of patients experiencing spontaneous remission, particularly those with germline diseases, such as Noonan or CBL syndrome, and some patients relapsing despite HSCT. Although there are shared clinical features with CMML, such as monocytosis and marked hepatosplenomegaly, the genetic landscape in JMML is distinct from adult myeloid neoplasms by the near absence of mutations in epigenetic and splicing modifiers. Up to 95% of children with JMML will possess a somatic or germline mutation in a Ras pathway gene (PTPN11, NF1, NRAS, KRAS, CBL).40-42 Despite some patients having identical genetic mutation profiles, differing clinical outcomes are observed. Recently, DNA-methylation patterns were shown to improve the prediction of outcomes, distinguishing JMML patients who experienced spontaneous remission from those who experienced an aggressive disease course.43
Other myeloid neoplasms with overlapping dysplastic and proliferative features
WHO-defined MDS/MPNs are considered distinct diagnoses, separate from the overlapping syndromes that they resemble. Yet myeloid malignancies can co-occur or have such nebulous boundaries that there exists an area of apparent diagnostic overlap. This can be challenging clinically, because treatment recommendations may differ across what may be rather arbitrary diagnostic borders. Consideration of clinical and molecular features may help to determine which condition should take precedence.
Due to its unique clinical and pathologic features, systemic mastocytosis (SM) is now recognized as its own disease category by the WHO. SM is divided into indolent SM (ISM), smoldering SM, SM with an associated clonal hematologic non–mast-cell –lineage disease that was renamed as systemic mastocytosis with associated hematologic neoplasm (SM-AHN) in the WHO 2016 update, aggressive SM, and mast cell leukemia.1 In addition to activating mutations in KIT, mutations in TET2, SRSF2, ASXL1, CBL, RUNX1, and RAS have been identified in patients with SM-AHN, aggressive SM, and mast cell leukemia.44 Additionally, mutations in ETNK1 are frequently seen in patients with SM with eosinophilia.45 Among patients with SM-AHN, these mutations may be coexpressed with KIT D816V in the same cells or expressed by other non–mast-cell myeloid cells.46,47 Colony assay studies found that KIT D816V mutations are often late events that are frequently preceded by mutations of TET2, SRSF2, and ASXL1, indicating that SM-AHN is a multimutated malignancy with diverging molecular evolution in subclones that have distinct differentiation potential.
Myeloid neoplasms account for 90% of all SM-AHN patients, including SM MPN (45%), SM CMML (29%), and SM MDS (23%).48 The largest study to date comparing patients with SM CMML (n = 50) vs CMML alone (n = 501) evaluated differences in clinical, cytogenetic, and genetic features, as well as clinical outcomes.49 Both groups had similar mutation profiles, with the exception of KIT and CBL mutations in the SM CMML cohort, suggesting that late KIT mutations may alter an initial CMML phenotype into one consistent with SM CMML.
Of note, KIT D816V may be viewed as a differentiation inducer in neoplastic cells rather than a dominant driver of oncogenesis, because patients with ISM express KIT D816V and do not typically have limited survival.50,51 Additional pathways mediated by oncogenic lesions preceding KIT mutations are likely responsible for a more aggressive disease phenotype, treatment resistance, and shortened survival. To this end, treatment of SM-AHM is focused on which disease component requires more immediate intervention. For example, a patient with an associated higher-risk CMML and resultant peripheral cytopenias may be treated with a hypomethylating agent, whereas mast cell–directed therapy may be appropriate for a patient with a lower-risk non–mast-cell malignancy and symptoms or organ dysfunction (“C findings”) related to the mast-cell component of the disease.52 Midostaurin is an approved tyrosine kinase inhibitor with activity against KIT D816V that demonstrated an overall response rate of 60% among patients with advanced SM.53 Additional studies are ongoing to evaluate alternative more selective KIT inhibitors. Future treatment strategies that extend beyond KIT are under investigation and include targeting pathways involving RAS, PI3K, mTOR, STAT5, and members of the BCL2 family.54,55
AA and hypoplastic MDS
Another area of diagnostic overlap occurs between AA and hypoplastic MDS (hMDS). Although the etiology of AA is typically considered distinct from that of MDS, with AA driven by immune-mediated destruction of hematopoietic stem/progenitor cells and MDS driven by a selective growth advantage of somatically mutated clonal hematopoietic stem/progenitor cells, in practice, these mechanisms may co-occur (Figure 1). First, among a subset of patients with lower-risk MDS, immune activation and inflammation drive the selection of somatically mutated clones, potentiating the response to immunosuppressive therapies (ISTs).56,57 Second, up to 15% of patients with severe AA (SAA) will have their disease evolve into MDS and/or acute myeloid leukemia (AML).58,59 Distinguishing AA from hMDS may be challenging, because patients with these diseases share many clinical features, such as bone marrow hypocellularity that hinders accurate evaluation of morphologic dysplasia, clonal cytogenetic and/or genetic abnormalities, and clinically meaningful responses to ISTs. Additionally, a subset of patients with AA harbor somatically mutated clones defined by mutations recurrently found in patients with MDS.60 A recent study evaluated somatic mutations in bone marrow samples from 150 patients with AA and no morphologic dysplasia.61 Excluding PIGA mutations, 29 of 150 (19%) patients harbored mutations, predominantly in ASXL1, DNMT3A, and BCOR (Figure 2). A total of 17 (11%) patients experienced progression to MDS, with 11 of these patients belonging to the group of 29 patients who possessed mutations. Somatic mutations were significantly associated with longer disease duration, shorter telomere lengths, and a greater likelihood of progressing to MDS or AML compared with patients without mutations. A similar study of 439 patients with AA found clonal hematopoiesis in 47% of patients, with inferior survival outcomes seen among patients with DNMT3A and ASXL1 mutations and higher IST response rates seen among patients with BCOR and PIGA mutations.62 SAA patients with MDS-like mutations were more likely to have these clones expand over time, particularly after IST. Other patients can harbor somatic copy number–neutral loss of heterozygosity at the HLA locus on chromosome arm 6p63 or mutations in HLAs and related pathways.64,65 These abnormalities appear to provide escape from HLA-restricted T-cell immunity driving SAA, occur more often in younger patients, and are associated with lower rates of neoplastic progression.66,67
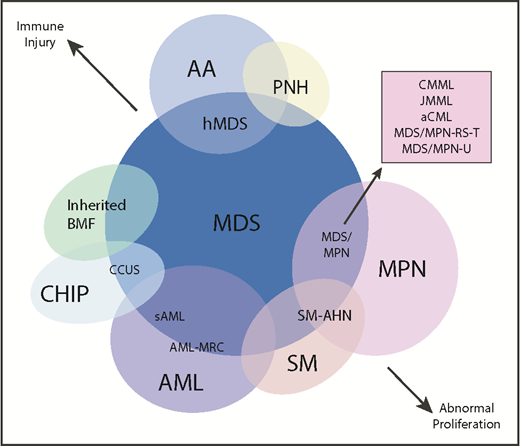
Diagram depicting myeloid disorders with clinical and genetic features shared with MDS and the degree to which they are driven by proliferative and immunologic mechanisms.
Diagram depicting myeloid disorders with clinical and genetic features shared with MDS and the degree to which they are driven by proliferative and immunologic mechanisms.
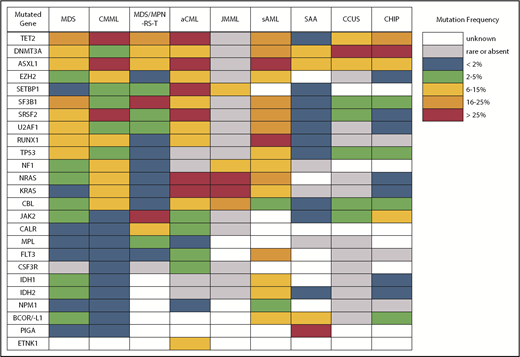
Differences in gene mutation frequency across different MDS/MPN overlap conditions and disorders at the diagnostic boundary with MDS.
Differences in gene mutation frequency across different MDS/MPN overlap conditions and disorders at the diagnostic boundary with MDS.
Approximately 15% to 20% of MDS bone marrows are hypocellular for age. These cases have differences in genetic profiles that include a lower rate of mutations and a lower frequency of splicing factor gene mutations compared with hyperplastic patients.68 This pattern is more similar to that observed in SAA. Because bone marrow cellularity is limited, morphologic dysplasia is difficult to evaluate when considering hMDS vs SAA or nonsevere AA. Other morphologic features outside of dysplasia that support a diagnosis of hMDS, or an MDS/MPN overlap syndrome, over AA include excess bone marrow blasts (≥2%), ring sideroblasts, extensive fibrosis, and circulating pseudo-Pelger–Huet cells. Certain cytogenetic abnormalities, such as del(5q), monosomy 7, and inversion 3, are considered presumptive evidence of MDS.69,70 The paucity of splicing factor and cohesin mutations in AA suggests that these lesions may also help to define the distinction between these disorders in the future. A lack of common MDS mutations or the presence of abnormalities of BCOR, PIGA, or the HLA loci correlate with more favorable outcomes in SAA and may be surrogate molecular markers of this disorder absent MDS defining features. In the meantime, a practical approach would be to minimize the distinction between hMDS and AA and simply consider patients at this boundary to be potentially responsive to immune suppression, reserving molecular studies to identify patients at risk for evolution to higher-risk disease.
One important caveat to this approach involves patients with inherited bone marrow failure syndromes, many of which can evolve into MDS or AML. For example, individuals with germline mutations of GATA2, DDX41, Fanconi anemia genes, or telomerase complex genes can have hypoplastic marrow findings well before the development of a clonal myeloid disorder that, in some cases, might never occur. Identifying these individuals is critical because their marrow failure does not respond to immune suppression. There are also important implications involving the health of family members, related stem cell donor candidates, and increased toxicity of IST or cytotoxic therapy. To make matters worse, some germline predisposition mutations, such as those in RUNX1 and ANKRD26, may cause thrombocytopenia and megakaryocyte dysplasia that could be mistaken for MDS-defining criteria.61,71 This diagnosis should not be made in the absence of other diagnostic elements.72 In this context, however, the presence of somatic mutations may indicate a greater risk for neoplastic progression.73,74 Mutation testing of presumed de novo MDS patients may also detect germline variants, because many of the genes tested are included in these panels.75 These variants can occur even in patients without a family history, young age of onset, or associated physical findings typical of germline predisposition syndromes.76 Dedicated testing of nonhematopoietic tissue is recommended in cases in which such a germline variant is suspected.77,78
Clonal hematopoiesis, unexplained cytopenias, and lower-risk MDS
Another diagnostic boundary with MDS involves patients with unexplained cytopenias often described as ICUS. These patients lack MDS-defining bone marrow criteria that include an increased blast proportion, specific cytogenetic abnormalities, or morphologic dysplasia in at least 10% of cells of a given lineage (Figure 3).79 Sequencing studies have identified somatic abnormalities indicative of clonal hematopoiesis in nearly 40% of ICUS patients, and closer to 70% in those who have some degree of dysplasia.80,81 These individuals are described as having a clonal cytopenia of undetermined significance (CCUS). Patients with CCUS can have many of the same mutated genes observed in lower-risk MDS and have comparable variant allele frequencies although mutations of SF3B1 appear to be more specific for MDS. Patients with a CCUS have a high rate of progression to MDS or other myeloid malignancies, particularly if they carry higher-risk features. These include somatic mutations in JAK2, RUNX1, any of the commonly mutated splicing factors (SF3B1, SRSF2, U2AF1, ZRSR2), or ≥2 somatically mutated myeloid malignancy genes.82 This risk may be as high as 90% at 5 years. For single mutations of DNMT3A, TET2, or ASXL1, the risk of progression is lower: ∼50% at 5 years. An absence of mutations on a broad panel of the 40 most frequently mutated MDS genes has a very low rate of progression approaching 1% per year of follow-up. Because MDS-defining bone marrow dysplasia can be hard to quantify, future revisions to MDS diagnostic criteria may include more clearly defined higher-risk CCUS patients, just as SF3B1 mutations are currently accepted as evidence of MDS-RS in patients with as few as 5% ring sideroblasts.1
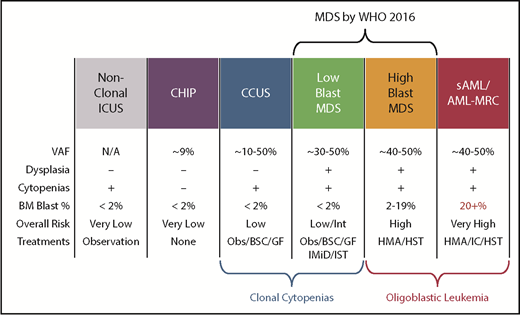
Comparison of features between cytopenic and clonal hematopoietic states that border MDS. BSC, best supportive care; GF, growth factors; HMA, hypomethylating agent; HST, hematopoietic stem cell transplant; IC, induction chemotherapy; IMiD, immunomodulatory imide drugs; Obs, observation.
Comparison of features between cytopenic and clonal hematopoietic states that border MDS. BSC, best supportive care; GF, growth factors; HMA, hypomethylating agent; HST, hematopoietic stem cell transplant; IC, induction chemotherapy; IMiD, immunomodulatory imide drugs; Obs, observation.
An important caveat is that somatic mutations typical of MDS can also occur in the blood cells of hematologically normal persons, with a prevalence that increases markedly with age.83,84 These individuals are said to have clonal hematopoiesis of indeterminate potential (CHIP) and, in the absence of cytopenias (or another concerning clinical context, such as a germline predisposition), are believed to have a very low risk for neoplastic progression (∼1% per year). CHIP mutations are most often found in DNMT3A, TET2, or ASXL1 (Figure 2) as isolated lesions with a low variant allele frequency (VAF) (<10%) and should not be considered diagnostic of MDS or any myeloid neoplasm. CHIP should also not be equated with CCUS, in which mutations are more frequent, of greater abundance, and associated with a much higher probability of malignant progression.80-82,85,86
MDS progression to sAML
At the other end of the prognostic spectrum for MDS lies the boundary with sAML. The border between these disorders has shifted over time, with the WHO classification for sAML currently defined as ≥20% bone marrow and/or peripheral blood blasts. Under the earlier French–American–British schema, MDS patients with 20% to 29% blasts were considered to have refractory anemia with excess blasts in transformation. The poor outcome of this latter group prompted the lower blast threshold set by the WHO; however, in retrospect, it is not clear that MDS patients with 10% to 19% bone marrow blasts have meaningfully different outcomes. In practice, these 2 groups straddling the divide between MDS and AML are treated in a similar fashion, receiving hypomethylating agents and considered for HSCT when appropriate. For these reasons, there have been calls to do away with the concepts of MDS with excess blasts and low-blast-count sAML, unifying them under the term “oligoblastic leukemia.”87 However, arbitrarily redefining the boundary between MDS and AML may not be enough. The challenge will be to identify those MDS patients who are headed toward leukemic progression and those who may have excess blasts but largely fail to progress.
The existence of the latter group can be inferred from the population of prognostically higher risk patients who live longer than the median for their revised International Prognostic Scoring System risk group.88 These individuals have a time-dependent risk that more closely resembles that of MDS patients with lower-risk disease. Because this determination is not made at diagnosis, several studies have attempted to risk-stratify MDS patients based on their leukemic potential earlier in the course of their disease. For example, Makishima et al examined tumor samples from >2000 patients for mutated genes enriched in higher-risk MDS and sAML.89 Mutations of NPM1, IDH1, IDH2, WT1, NRAS, PTPN11, and FLT3 were found significantly more often in the sAML cohort. Mutations of these genes were typically subclonal to a more abundant mutation (suggesting that they were acquired later) and were associated with significantly shorter progression-free survival. In MDS, acquisition of these gene mutations may define leukemic clones that might not lead to a clinical definition of sAML for many months. Such patients could be said to harbor an overlap disorder between MDS and sAML. One implication of this hypothesis is that therapies targeted at these subclones (eg, IDH or FLT3 inhibitors) may not lead to traditionally defined hematologic responses but, nonetheless, may delay leukemic transformation. This prediction will have to be tested in prospective clinical trials.
Patterns of gene expression have also been used to identify MDS patients at greatest risk of leukemic progression. Shiozawa et al examined the transcriptomes of CD34+ bone marrow cells from patients with MDS.90 Unsupervised clustering identified 2 major subgroups: 1 enriched for the expression of genes associated with erythroid and megakaryocytic differentiation and another defined by transcripts associated with immature progenitors (IMPs). The erythroid and megakaryocytic differentiation subgroup had longer overall survival and was associated with SF3B1 mutations, ring sideroblasts, and a strong erythroid signature. In contrast, the IMP subgroup had lower platelet counts, increased marrow blasts, and higher-risk mutations. Strikingly, only patients in the IMP subgroup had disease that transformed into sAML, suggesting that, even in the absence of “leukemic” mutations, some forms of MDS have greater leukemic potential that can be recognized well before progression takes place.
The second area of overlap between MDS and AML involves patients who may not have had a recognized antecedent MDS but are diagnosed with AML with myelodysplasia-related changes (AML-MRC), suggesting a pathogenic link with MDS.91,92 Molecular profiling may help to segregate those with MDS and AML-MRC from those with de novo AML, providing prognostic information for the patient and clinician. These patients will frequently harbor MDS-associated cytogenetic abnormalities and are often classified as having higher-risk disease. Not surprisingly, patients with AML-MRC are more likely to harbor mutated genes typical of MDS and sAML, including splicing factors (SRSF2, SF3B1, and U2AF1), chromatin modifiers (EZH2 and ASXL1), and STAG2 and BCOR (Figure 2).93,94 Older individuals with AML are more likely to carry somatic mutations in these genes, even if they are not described as having AML-MRC. Importantly, older de novo AML patients without these mutations have a more favorable response to therapy and duration of remission, making it important to identify them at diagnosis.
From another perspective, one could consider MDS with excess blasts to be an overlap syndrome between lower-risk MDS defined by clonal cytopenias with bone marrow failure and oligoblastic myeloid leukemia (Figure 3).86,95 In practice, patients with higher-risk MDS or low-blast-count AML have a similar prognosis and are often treated with hypomethylating agents or, if appropriate, considered for stem cell transplantation.96 Altering our diagnostic boundaries between MDS and AML based on underlying mutations and clinical phenotypes may more accurately classify patients with these conditions.
Conclusions
MDS overlap syndromes are genetically and clinically heterogeneous disorders that can represent distinct biological entities or areas of diagnostic ambiguity. Although WHO-defined disease classifications rely largely on morphologic criteria, molecular markers of disease are increasingly able to identify differences in clinical phenotypes when considered in the appropriate clinical context. There are no specific mutations that stringently diagnose MDS overlap syndromes or unequivocally define diagnostic boundaries with MDS; however, future classification schemes are sure to incorporate our growing understanding of the molecular basis of these disorders.
Authorship
Contribution: T.N.T. and R.B. cowrote this review and edited the submitted manuscript.
Conflict-of-interest disclosure: R.B. has served as a consultant for Celgene and Genoptix, has received research funding from Celgene and Takeda, has served on a data safety monitoring board for Celgene-sponsored clinical trials, and has received honoraria for speaking at education conferences sponsored by Celgene and Xian Janssen. T.N.T.declares no competing financial interests.
Correspondence: Rafael Bejar, University of California San Diego Moores Cancer Center, 3855 Health Sciences Dr, MC 0820, La Jolla, CA 92093-0820; e-mail: rabejar@ucsd.edu.
