

Abstract
Juvenile myelomonocytic leukemia (JMML) is a unique clonal hematopoietic disorder of early childhood. It is classified as an overlap myeloproliferative/myelodysplastic neoplasm by the World Health Organization and shares some features with chronic myelomonocytic leukemia in adults. JMML pathobiology is characterized by constitutive activation of the Ras signal transduction pathway. About 90% of patients harbor molecular alterations in 1 of 5 genes (PTPN11, NRAS, KRAS, NF1, or CBL), which define genetically and clinically distinct subtypes. Three of these subtypes, PTPN11-, NRAS-, and KRAS-mutated JMML, are characterized by heterozygous somatic gain-of-function mutations in nonsyndromic children, whereas 2 subtypes, JMML in neurofibromatosis type 1 and JMML in children with CBL syndrome, are defined by germline Ras disease and acquired biallelic inactivation of the respective genes in hematopoietic cells. The clinical course of the disease varies widely and can in part be predicted by age, level of hemoglobin F, and platelet count. The majority of children require allogeneic hematopoietic stem cell transplantation for long-term leukemia-free survival, but the disease will eventually resolve spontaneously in ∼15% of patients, rendering the prospective identification of these cases a clinical necessity. Most recently, genome-wide DNA methylation profiles identified distinct methylation signatures correlating with clinical and genetic features and highly predictive for outcome. Understanding the genomic and epigenomic basis of JMML will not only greatly improve precise decision making but also be fundamental for drug development and future collaborative trials.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a clonal myeloproliferative/myelodysplastic neoplasia characterized by constitutive activation of the Ras signal transduction pathway. Canonical Ras pathway mutations in the PTPN11, NRAS, KRAS, NF1, or CBL genes are present in leukemic cells of∼90% of patients. Despite major advances in molecular diagnostics, JMML remains a puzzling disorder with diverse natural history and outcome. The complexity of the entity is in part due to the observation that mutations in the respective genes can either occur as germline (“syndromic”) or as somatic lesion in hematopoietic cells (“nonsyndromic”). Furthermore, within a genetically defined subtype, the clinical course varies widely depending on the presence of clinical risk factors like age, hemoglobin F (HbF), thrombocytopenia, or the more recently defined methylation classes. Thus, appropriate clinical management for JMML patients ranges from watchful observation to early allogeneic hematopoietic stem cell transplantation (HSCT). In this review, we focus on the genetic and epigenetic features of JMML and outline their impact on clinical care.
JMML nosology in a nutshell: a historical appraisal
Guidance of therapy decisions for a child with JMML requires a detailed understanding of JMML pathobiology. Appreciation of some landmarks in JMML research might ease the grasp of current concepts (Figure 1).
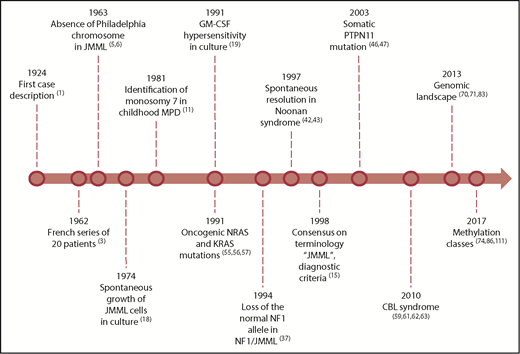
From the first case descriptions to prognostic factors
The first case report of JMML was published in 1924,1 followed by additional case descriptions and small series of children with chronic granulocytic leukemia.2 In 1962, Bernard and coworkers were the first to carefully describe a larger series of 20 infants with myelomonocytic leukemia.3 They pointed to the young age of these children, male predominance, splenomegaly in virtually all patients, as well as leukocytosis with precursors on smear, and monocytosis. Moreover, they saw cases with underlying neurofibromatosis type 1 (NF1), and an occasional infant with unexplained spontaneous resolution, and subsequently identified prognostic factors for survival.4
Following the discovery of the Philadelphia chromosome in 1960, the clinical and hematological picture of JMML was contrasted with Philadelphia chromosome–positive chronic myeloid leukemia.5,6 In addition, greatly raised HbF levels6-8 and the presence of other fetal red cell characteristics6,8,9 were identified as characteristic features of this disorder.
Since the first reports of a missing group C chromosome in children with myeloproliferative disorders (MPD),10 childhood monosomy 7 was perceived as a separate entity in the 1980s.11,12 An infantile monosomy 7 syndrome with clinical features similar to JMML, but low HbF levels, was proposed.13 The retrospective series of the European Working Group of Myelodysplastic Syndromes (MDS) in Childhood (EWOG-MDS) of 110 patients14 confirmed that low platelet count, age ≥2 years, and high HbF at diagnosis are the main clinical predictors of poor survival.4,13,14 To clear up a clumsy nosology, in 1996 an international working group introduced the term JMML and established criteria for its diagnosis.15 The World Health Organization classification placed the entity in the group of mixed myelodysplastic/MPD.16,17
JMML hematopoiesis in cell culture and clonality
The introduction of culture methods for hematopoietic progenitor cells in the 1970s allowed studying the proliferative properties of peripheral blood (PB) and bone marrow (BM) cells of children with JMML. When cultured in semisolid media, JMML cells give rise to an excess number of monocyte-macrophage colonies in the absence of added growth factors.18 This so-called spontaneous proliferation of JMML myeloid progenitor cells depends on an endogenous production of cytokines by monocytes, but is primarily due to a striking hypersensitivity of progenitors to granulocyte-macrophage colony stimulating factor (GM-CSF) in vitro.19 Although not completely specific to JMML, GM-CSF in vitro hypersensitivity became a hallmark of the disease and an important diagnostic tool. GM-CSF was shown to be obligatory for survival of JMML cells,20,21 while the role of other cytokines like interleukin-122 or tumor necrosis factor-α23 remained somewhat controversial. Advanced phospho-specific flow cytometry was later used to assess STAT5 activation as a parameter for pathway activity without the need for time-consuming cell cultures.24,25
Culture studies also provided the first evidence that erythroid progenitor cells participate in the neoplastic clone,6 a finding later substantiated by X-chromosome inactivation patterns12 and genetic markers.12,26,27 Molecular studies in JMML patients with B-lineage blastic transformation28,29 or concurrent30,31 or consecutive32,33 T-cell precursor lymphoid neoplasia provided further clinical evidence that JMML arises from a multipotent stem cell.
RASopathies and somatic drivers
Predisposition to JMML in children with NF1 was evident from the early clinical descriptions.3,34 Although JMML is an uncommon complication of NF1, the risk of developing JMML for the patient with NF1 is estimated 200- to 350-fold higher than in patients without NF1.14,35 In 1990, the NF1 gene was cloned, and neurofibromin, the encoded gene product, was shown to function as GTPase activating protein, accelerating the intrinsic rate of Ras-GTP hydrolysis to the inactive form, Ras-GDP.36 Four years later, Shannon et al demonstrated the loss of the wild-type NF1 allele in BM cells of children with NF1 and JMML, thus establishing NF1 as a tumor suppressor gene.37 JMML cells from children with NF1 showed a selective decrease of NF1–GTPase activating protein activity as well as elevated levels of Ras bound to GTP.38 Subsequent genetic studies indicated that loss of heterozygosity at the NF1 locus was predominantly due to segmental uniparental disomy (UPD) of large parts of chromosome 17q.39-41
When in 1997 French42 and Japanese43 investigators indicated that some of the children with JMML had underlying Noonan syndrome (NS), a second RASopathy was implicated in MPD of infancy. In contrast to NF1-associated JMML, NS/MPD generally resolves spontaneously over months or years.42-44 The discovery of heterozygous germline gain-of-function PTPN11 mutations as the major cause of NS45 and subsequently in NS with transient MPD46 led to the prediction that children with “nonsyndromic” JMML may bear somatic PTPN11 mutations in their leukemic cells. In fact, PTPN11, the gene encoding nonreceptor tyrosine phosphatase SHP2, was found to be the most commonly mutated gene in JMML46,47 (Figure 2).
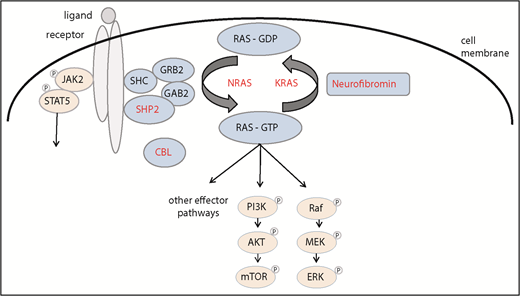
The spectrum of PTPN11 mutations observed in patients with JMML, NS/MPD, and NS suggested a genotype/phenotype correlation with germline PTPN11 mutations predicted to result in a weaker gain of function than the somatic alterations.44,48 NS proved to be genetically heterogeneous with a spectrum of underlying Ras pathway alterations, including KRAS49 and NRAS.50 Analogous to the dual role of PTPN11 as both oncogene and developmental gene, NS-associated KRAS mutations were shown to have milder biochemical effects than the typical oncogenic somatic mutations,49 offering an explanation as to why these lesions are tolerated during embryonic development. Also, similar to what had been observed in PTPN11-mutated NS, a transient MPD was noted in some children with NS and KRAS,51 NRAS,52 or RIT153 germline mutations.
Appreciating the fundamental role of Ras as master switch of cellular proliferation, differentiation, and survival, heterozygous somatic gain-of-function mutations in RAS genes were the first specific genetic alterations identified in human cancer in the early 1980s.54 In JMML, oncogenic mutations in NRAS or KRAS were detected in hematopoietic cells of JMML patients without NF1,27,55-58 consistent with the hypothesis that 1 activating Ras pathway mutation would be sufficient to cause JMML.57
When high-density single nucleotide polymorphism microarrays allowed the systematic detection of regions of copy number gains and losses, we noted UPD 11q in BM cells of children with JMML negative for mutations in PTPN11, NRAS, KRAS, or NF1.59 Similar observations were made in adult MPD, and subsequent analysis identified homozygous missense mutations in the CBL gene encoding for a RING finger ubiquitin ligase and multiadapter protein.60 Based on these observations, we identified homozygous or, rarely, heterozygous CBL mutations in ∼10% of patients with JMML.59,61 Many of these children were known to have syndromic features. In fact, in contrast to adult MPD, children with JMML and CBL mutations were found to have germline CBL mutations.62,63 CBL-syndrome, a previously unreported RASopathy implicated in JMML, shares many features with mild forms of NS. Although the mechanism of leukemic development (acquired biallelic inactivation of the gene after monoallelic alteration in the germline) is the same in NF1- and CBL-associated JMML, the natural course of the 2 genetic JMML subtypes was found to be very different. In contrast to patients with NF1-associated disease, many of the children with CBL-mutated JMML experience spontaneous regression of myeloproliferation.62,63
The clinical and hematologic JMML phenotype
The most consistent features of the JMML phenotype are young patient age, splenomegaly, presence of myelocytes, metamyelocytes, and often nucleated red cells on PB smear, and a BM aspirate with a normal or only moderately increased blast count.14 An elevated HbF level is supportive of the diagnosis. These features are generally sufficient to trigger molecular diagnostic studies for JMML.
In most children with JMML, leukemic infiltrates of spleen, liver, and lung are clinically obvious. In rare patients without splenomegaly, genetic studies can confirm the diagnosis. Hepatomegaly is generally less prominent than splenomegaly. Dry cough, tachypnea, and interstitial infiltrates on chest radiograph may be indistinguishable from respiratory infections. Gastrointestinal infiltration can result in diarrhea and in rare cases in gut perforation. Leukemic skin lesions are pleomorphic ranging from eczematous eruptions (cradle cap) to indurated raised lesions with central clearing to Sweet syndrome. In addition, nonspecific lesions like juvenile xanthogranuloma may be present. Although 1 to 2 café-au-lait spots are often seen in patients with CBL syndrome,62 the clinical diagnosis of NF1 in infants with JMML requires ≥6 lesions of >5 mm in size. Because half of the children with JMML and NF1 inherit their genetic predisposition, the value of taking a family history (and if appropriate inspection of mother’s or father’s suspicious skin lesions) cannot be overestimated. In our own experience, close to all children with JMML and underlying NF1 can be diagnosed clinically. Likewise, especially in young infants with JMML, phenotype should be carefully examined for the presence of typical features of NS, such as facial dysmorphia, heart disease, failure to thrive, hearing loss, and others.
Leukocytosis is common in JMML, but a presenting white blood count <10 × 109/L is occasionally noted.13,14 In addition to immature granulocytes and erythroid precursors, a few blasts may be present; the median blast percentage in PB is <2%.4,14 Although most cases show a striking monocytosis, often with dysplastic forms, the absolute monocyte count can be <1 × 109/L,13,14 a threshold applied as lower limit in previous diagnostic algorithms.15,17,64,65 The vast majority of JMML patients have thrombocytopenia with the exception of children with NF1-associated JMML, who show platelet counts within the normal range in most cases.14 Anemia is generally not a leading symptom and rarely requires red blood cell (RBC) transfusion. Although RBCs are most often normocytic, macrocytosis is noted in some patients with monosomy 7,14 and occasional cases with persistent microcytosis66 might be due to epigenetic dysregulation of the β-like globin genes.67 BM examination is required to exclude acute leukemia. In JMML, BM findings are not by themselves diagnostic but rather compatible with the diagnosis; the most consistent finding in BM specimens is the reduced number of megakaryocytes.14 Cytogenetic studies of JMML cells show a normal karyotype in 65% of cases, sole monosomy 7 in ∼ 25%, and other aberrations in 10%.14 The likelihood of an abnormal karyotype is dependent on the genetic subtype, with monosomy 7 being noted most often in KRAS-mutated disease (Figure 3).
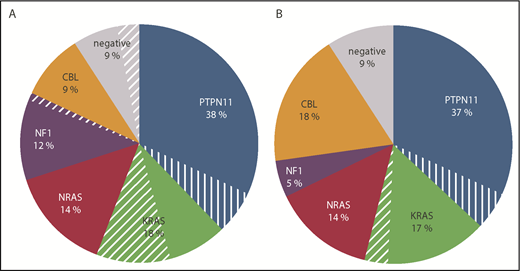
Distribution of Ras pathway mutations in children with JMML. Data were reported by (A) the EWOG-MDS (N = 142)86 or (B) the Japanese JMML Cooperative Study Group (N = 132).74 The 3 cases of the Japanese cohort with ALK or ROS1 rearrangement had been excluded from the analysis. Cases with monosomy 7 are indicated by the hatched slices. The analyses do not include patients with NS/MPD.
Distribution of Ras pathway mutations in children with JMML. Data were reported by (A) the EWOG-MDS (N = 142)86 or (B) the Japanese JMML Cooperative Study Group (N = 132).74 The 3 cases of the Japanese cohort with ALK or ROS1 rearrangement had been excluded from the analysis. Cases with monosomy 7 are indicated by the hatched slices. The analyses do not include patients with NS/MPD.
Earlier investigations noted that the majority of children with JMML have elevated immunoglobulin G (IgG), IgM, and IgA levels.4,14,68 Like in the RASopathies,69 autoantibodies (such as antinuclear antibodies, antibodies against RBCs giving rise to a positive antiglobin test, or antithyroglobulin antibodies) can be present in JMML but rarely give rise to clinical symptoms.
Diagnostic procedures and differential diagnosis of the JMML phenotype
Between subtypes, the clinical and hematological genotype-phenotype correlation is poor with the exception of syndromic features, like facial phenotype, presence of heart disease, or presence of café-au-lait spots in children with RASopathies. NF1 in JMML patients can be diagnosed clinically, and mutational analysis of the PTPN11, NRAS, KRAS, and CBL gene in hematopoietic and nonhematopoietic tissue can provide an unequivocal diagnosis in the vast majority of JMML patients. Nonhematopoietic tissue is often easily obtained from hair follicles; buccal swabs are prone to contamination but can be helpful when negative for the Ras pathway mutation found in PB/BM. Timely parental counseling and therapeutic guidance can be greatly facilitated by providing reference laboratories simultaneously with hematopoietic and nonhematopoietic tissue after the appropriate genetic consent had been obtained.
Results from cooperative study groups have demonstrated that the PTPN11-, NRAS-, KRAS-, CBL-, and NF1-associated subtypes account for ∼38%, 18%, 14%, 12% to 18%, and 5% to 10% of JMML patients, respectively (Figure 3). In a few of the cases negative for all 5 canonical mutations, activating somatic RRAS mutations have been described.70,71 Mutations in this small GTPase with 50% to 60% homology to the RAS-proteins72 result in an atypical phenotype with rapid clinical progression to acute myeloid leukemia (AML).
The differential diagnosis of the JMML phenotype includes rare myeloproliferative malignancies with receptor tyrosine kinase translocations. Identification of these cases is crucial, because patients may benefit from receptor tyrosine kinase–targeted inhibitors. In some patients, ALK receptor rearrangements, often associated with monosomy 7, were described.73,74 Ras pathway mutation-negative JMML with increased eosinophils needs to be differentiated from MPD with eosinophilia and constitutively activated platelet-derived growth factor receptor α or β, or fibroblast growth factor receptor 1.75,76 Furthermore, infants with KMT2A rearrangements can occasionally present with hepatosplenomegaly and low blast count.77,78
Among the nonneoplastic disorders, infections are usually evident. Wiskott-Aldrich syndrome may need to be considered in male infants,79 whereas leukocyte adhesion deficiency generally does not give rise to thrombocytopenia.80 Infantile malignant osteopetrosis (IMO) can mimic all clinical and hematological features of JMML; radiographic imaging studies demonstrating increased bone density are helpful in distinguishing IMO from JMML.81
JMML genetic subtypes and therapeutic considerations
Depending on the genetic subtype, therapeutic considerations for children with JMML range from observation to allogeneic HSCT. Age ≥2years, severe thrombocytopenia and/or a high HbF level indicate aggressive disease with high risk for relapse after HSCT.15,82 The features of the genetic subtypes discussed below are summarized in Table 1.
Genetic subtypes of JMML
| I. Somatic PTPN11 mutation |
| • Rapidly fatal without allogeneic HSCT |
| • High probability of relapse |
| • Frequent acquisition of NF1 haploinsufficiency |
| II. Somatic NRAS mutation |
| • Heterogeneous subtype |
| • Rapid progress with high relapse rate after HSCT, typically in older children with high levels of HbF |
| • Indolent course with spontaneous regression, typically in infants or in cases with G12S mutation |
| III. Somatic KRAS mutation |
| • Mostly infants |
| • Frequent association with monosomy 7 |
| • Aggressive at presentation but low risk of relapse after allogeneic HSCT |
| IV. JMML in children with NF1 |
| • Older age at diagnosis |
| • Higher platelet count |
| • Higher percentage of BM blasts |
| • Fatal without allogeneic HSCT |
| V. JMML in children with germline CBL mutation |
| • Loss of CBL heterozygosity in hematopoietic cells |
| • Absence of concomitant mutations |
| • Value of allogeneic HSCT uncertain |
| • Frequent occurrence of mixed chimerism after allogeneic HSCT |
| I. Somatic PTPN11 mutation |
| • Rapidly fatal without allogeneic HSCT |
| • High probability of relapse |
| • Frequent acquisition of NF1 haploinsufficiency |
| II. Somatic NRAS mutation |
| • Heterogeneous subtype |
| • Rapid progress with high relapse rate after HSCT, typically in older children with high levels of HbF |
| • Indolent course with spontaneous regression, typically in infants or in cases with G12S mutation |
| III. Somatic KRAS mutation |
| • Mostly infants |
| • Frequent association with monosomy 7 |
| • Aggressive at presentation but low risk of relapse after allogeneic HSCT |
| IV. JMML in children with NF1 |
| • Older age at diagnosis |
| • Higher platelet count |
| • Higher percentage of BM blasts |
| • Fatal without allogeneic HSCT |
| V. JMML in children with germline CBL mutation |
| • Loss of CBL heterozygosity in hematopoietic cells |
| • Absence of concomitant mutations |
| • Value of allogeneic HSCT uncertain |
| • Frequent occurrence of mixed chimerism after allogeneic HSCT |
PTPN11-mutated JMML
All somatic PTPN11 alterations in JMML are missense mutations in the N-terminal SH-2 (exon 3) or PTP-interacting surfaces (exon 13) and result in gain of function.46,47 Acquisition of NF1 haploinsufficiency is a frequent subclonal event.70,71,74,83 JMML with PTPN11 mutation is a rapidly fatal disorder unless allogeneic HSCT can successfully be performed.65 In some HSCT series, patients with PTPN11 mutations had a significantly worse outcome with higher relapse rates when compared with patients with the other JMML genetic subtypes.84,85
NRAS-mutated JMML
Among the typical cancer-associated somatic NRAS mutations at codons 12, 13, and 61, acquisition of a G12D, G13D, or G12S allele is the most frequent change.70,86 A considerable percentage of patients with NRAS-mutated JMML relapse after HSCT,65,87 but some young infants survive in the absence of HSCT with persistence of the oncogenic NRAS mutation and slowly regressing disease.88-90 Clinically, these children are well and show a normal or only slightly elevated HbF.
KRAS-mutated JMML
Most children with somatic heterozygous KRAS mutations are diagnosed below the age of 1 year.91 They often present with particularly severe disease. Monosomy 7 is frequently noted in leukemic cells70,86 (Figure 3); the significance of this observation remains elusive. In some cases of KRAS-mutated JMML, an impressive treatment response to azacitidine has been observed.92,93 Patients with KRAS-mutated JMML transplanted after a preparative regimen with busulfan, cyclophosphamide, and melphalan have a low relapse rate after allogeneic HSCT and may benefit from less intensive preparative regimens.65 KRAS-mutated JMML shares many features with a rare condition called RAS-associated lymphoproliferative disease.94,95 The 2 entities may represent different phenotypes of the same disorder.96
NF1-mutated JMML
Although in the majority of patients biallelic NF1 gene inactivation is due to UPD, compound-heterozygous NF1-inactivating mutations and occasionally somatic interstitial deletions account for up to a third of the cases.39,41 Children with JMML and NF1 have a higher platelet count, have a higher percentage of blasts in BM, and are more often diagnosed after the age of 5 years than JMML patients of other subtypes.14 Although some of the younger children can initially enjoy a relatively unaffected clinical course, NF1-mutated JMML is invariably fatal unless allogeneic HSCT is successful.65
CBL-mutated JMML
Germline mutations in children with CBL-mutated JMML are located throughout the linker and RING finger domain (intron 7, exons 8 and 9) of the CBL gene.59 Most patients have 11q isodisomy in hematopoietic cells, and heterozygous mutations have been reported in a few cases.59,62 Secondary genetic alterations are conspicuously absent.70,71,83 Most children with CBL-mutated JMML have self-limiting disease with persistence of clonal hematopoiesis.62,63 Observation without therapeutic intervention is generally advised, but in some instances grossly enlarged spleens and thrombocytopenia require therapeutic intervention. In addition to JMML, patients with germline CBL mutations have a high risk for vasculopathy and neurological disease.59,62,97,98 Clinical observations97,98 and animal models99 suggest that these pathologies are mediated, at least in part, by CBL-deficient T lymphocytes and might therefore be amenable to prevention by allogeneic HSCT.
JMML without known Ras pathway mutation
In children with a JMML phenotype but absence of a known Ras pathway mutation, other rare MPD, acute leukemia, and some benign disorders like IMO need to be excluded (see “Diagnostic procedures and differential diagnosis of the JMML phenotype”). Molecular analysis of the NF1 gene and of secondary mutations typically noted in JMML may be helpful to confirm the clinical diagnosis of JMML.
Secondary genetic alterations
Several publications on the genetic landscape of JMML have documented secondary mutational events occurring inside or outside the canonical Ras pathway axis, including RAS double mutants, components of the polycomb repressive complex 2 (like EZH2 and ASXL1), SETBP1, JAK3, and occasionally, spliceosome genes. It appears that such mutations characterize patients with the highest risk of progression and poor outcome.70,71,74,83 Interestingly, recent evidence suggests that clones carrying these mutations are often minuscule at diagnosis but preferentially expand at the time of relapse after HSCT.87 Other authors linked differential expression of key regulatory noncoding RNAs, such as let-7100 or miR-150-5p,101 to the various genetic subgroups of JMML.
DNA methylation classes
The canonical genetic subtypes of JMML do not fully explain the diverse nature of JMML. The known clinical risk factors of age, HbF, and platelet count13,14,82 are only weakly associated with the type of index mutation. In addition, several molecular features like AML-like gene-expression pattern,102 occurrence of genetic comutations83,87 or deregulation of the fetal hematopoietic regulator gene LIN28B100 were identified and shown to have strong predictive power, but again cannot be directly coupled to the genetic subtypes. From these considerations arose interest to study epigenetic processes in JMML, in particular, the dysregulation of genomic DNA methylation, which is a crucial component of Ras-driven malignant cell transformation.103
Investigators from EWOG-MDS used high-throughput mass spectrometry for the quantification of cytosine guanine dinucleotide (CpG) methylation of 15 candidate DNA regions in samples from 127 patients with JMML and thus were the first to identify CpG island hypermethylation of specific target genes as a recurrent feature of JMML cells.104 DNA hypermethylation was only weakly associated with the canonical genotypes or cytogenetic aberrations. Instead, it correlated strongly with classical parameters predictive of aggressive disease progression and poor outcome, especially older age and increased HbF level. The study suggested that methylation classes in JMML were not dichotomous, but were better represented in a tripartite fashion (characterized by moderate, intermediate, and high hypermethylation). Not surprisingly, the relationship between prognostic features and DNA methylation clearly translated into 1 between methylation and survival. The 5-year probability of survival was 72% in patients with low methylation, but 41% in those with high methylation. High methylation characterized a group of cases with high probability of relapse after HSCT, with the 5-year cumulative incidence of relapse being 52% vs 10% in low methylation. About three-quarters of the studied regions (although selected for reported hypermethylation in other malignant myeloid diseases) were not at all affected by aberrant methylation, highlighting that these processes are not uncontrolled random events in the transformed cell, but more likely an integral part of the specific cancer phenotype.
A series of subsequent studies examining additional candidate genes confirmed the essential findings of Olk-Batz et al,67,104-108 collectively showing that hypermethylation targeting different genes was highly correlated within individual JMML samples. Together with the fact that the relationship to prognosis was consistent regardless of the specific target region considered, this suggested the existence of a common CpG island methylator phenotype in JMML, similar to observations in colon cancer or poor-risk neuroblastoma.109,110
With the advent of array-based technology for the genome-wide interrogation of CpG methylation, 3 international groups independently embarked on large-scale investigations of JMML samples.74,86,111 Analyzing 167 cases, the EWOG-MDS demonstrated that 3 distinct classes of methylation patterns emerged when the 5000 CpG dinucleotides with the highest variation between the JMML samples were subjected to unsupervised cluster analysis.86 Confirming the earlier candidate-gene findings, these categories were characterized by low, intermediate, and high DNA methylation. The low-methylation cluster comprised infants with NRAS mutation, patients with CBL syndrome, and children with NS and transient MPD, thus patients known to have a favorable prognosis (Figure 4). A surprising and remarkable association was that of KRAS mutation and monosomy 7 with the intermediate cluster. The high-methylation group was dominated by older children and cases with PTPN11 mutation, resulting in poor outcome. Hinting at possible functional links between Ras activation and methylation classes, the authors also reported that DNA hypermethylation in JMML was more pronounced when additional mutations in Ras pathway genes or epigenetic modifier genes were present or DNA methyltransferases DNMT1 and DNMT3B were expressed at higher levels. Investigators in North America profiled 39 children diagnosed with JMML and reported that DNA methylation clusters defined 3 groups of patients with significant difference in event-free survival.111 The authors used samples from the EWOG-MDS to independently validate the signature. They also suggested that a certain CpG methylation profile, which resembles healthy leukocytes more than other JMML cases, might be capable of predicting spontaneous resolution of the disease. A group in Japan used cell samples from 106 children with JMML and defined 2 categories of DNA methylation.74 As in the previous 2 studies, the hypermethylation profile corresponded with JMML risk factors and poor prognosis. In addition, it was associated with a higher number of gene mutations, overexpression of LIN28B, and AML-like gene-expression profile.
![Figure 4. Methylation classes in JMML. Unsupervised cluster analysis of 1000 most variably methylated CpG dinucleotides (represented in rows with the methylation values coded in shades of color from blue [0%] to red [100%]) in 147 patients (columns) identifies 3 classes with characteristic differences in the distribution of Ras pathway mutations (genotype) and probability of relapse after HSCT. Adapted from Lipka et al86 with permission.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/10/10.1182_blood-2018-11-844688/3/m_blood844688f4.png?Expires=1769844265&Signature=3Xx5qzjLXg2~gdLqAekJuyNgOhf19q4aRmOB0Ia2YIMod~tbjG0rdSiO4mB3q4nSPFhCU-23QOtJ5i9pAzvC-~dkgQOdzX2DmWHUUgKVaKlXLMILnOqpoIzwHA6lwRBBFB8KGlhUjZaiqBm1r3H6zQFVCQW4fVMaopesBmAChLVQeF1yyWP~bOeV1eGdBF843Aer4Nje7ud6KXnLEMLv-5fM7-qMKkSvck-t1RfkqZCeCUf6IsW2WLLdy0w~F3NwZqRVDmtoQ8ILrI7xBeWCEjO79FdrCDyT2zVLPLI2pn4uYISkpb~oGiRSSSreF4ZZfpTCFjklTuQaKB4pGRmY4Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Methylation classes in JMML. Unsupervised cluster analysis of 1000 most variably methylated CpG dinucleotides (represented in rows with the methylation values coded in shades of color from blue [0%] to red [100%]) in 147 patients (columns) identifies 3 classes with characteristic differences in the distribution of Ras pathway mutations (genotype) and probability of relapse after HSCT. Adapted from Lipka et al86 with permission.
Methylation classes in JMML. Unsupervised cluster analysis of 1000 most variably methylated CpG dinucleotides (represented in rows with the methylation values coded in shades of color from blue [0%] to red [100%]) in 147 patients (columns) identifies 3 classes with characteristic differences in the distribution of Ras pathway mutations (genotype) and probability of relapse after HSCT. Adapted from Lipka et al86 with permission.
Taken together, the results of all DNA methylome studies in JMML were exceptionally consistent, and it can be expected that the methylation classes will supplement the diagnostic and prognostic parameters used previously. Future research will be directed at better understanding the mechanistic link of epigenetic dysregulation and resistance to treatment, for example, via harnessing epigenomic information for an improved definition of the “cell of origin” of JMML.112
Disease models
From the very beginning, the research community faced the problem that JMML eludes easy in vitro modeling. Longer-term preservation of primary patient-derived cell samples in suspension culture does not usually succeed because of rapid differentiation and senescence. An immortalized cell line, which accurately reflects the lineage diversity of JMML, has not been generated so far. Clonogenic cultures in semisolid methylcellulose medium had been instrumental in elucidating the proliferative mechanism of JMML, particularly GM-CSF hypersensitivity,19,22 and had provided crucial clues for the identification of the paradigmatic role of Ras signal transduction in JMML.113 Investigators then replicated oncogenic lesions of the Ras signaling pathway in genetically engineered mouse strains that encompassed transgenic germ line mutations114 or deletions115,116 as well as hematopoiesis-specific inducible alleles.117-119 Collectively, these models demonstrated that introduction of JMML-specific mutations is sufficient to induce an MPD resembling the clinical and proliferative properties of JMML. Two current possibilities to employ original JMML cells for preclinical research are xenotransplantation into immunodeficient recipient mice and induced pluripotent stem cell (iPSC) culture. The former was pioneered in experiments demonstrating that JMML cells engraft severe combined immunodeficient (SCID) mice120 or nonobese diabetic/SCID mice121 and that those JMML-initiating cells have sufficient self-renewal capacity to repopulate secondary murine recipients.120 The nonobese diabetic–SCID xenograft models were further enhanced by an additional interleukin-2 receptor γ chain deletion to eliminate residual natural killer cell activity,122 and transgenic expression of human cytokines.123 More recently, xenotransplantation of JMML cells into the Rag2−/−γc−/− mouse strain was described to result in long-term engraftment without the need for exogenous supply of GM-CSF, expansion of leukemic cell material outside the human organism, and successful retransplantation.124 The authors used the Rag2−/−γc−/− system to model therapy with the DNA methyltransferase inhibitor 5-azacytidine and reported preferential depletion of the CD34+ JMML progenitor cell pool after treatment with 5-azacytidine compared with cytosine arabinoside.125 In addition, they showed that leukemic DNA methylation profiles were reestablished in the xenograft, indicating their origin in the leukemia-initiating cell.125 To create a renewable source of cells for research on JMML, researchers in Philadelphia used lentiviral transduction of the 4 Yamanaka factors into material from 2 children with PTPN11 p.E76K JMML and succeeded in establishing iPSCs with phenotypic and functional qualities resembling original JMML cells.126 Recently, the authors contrasted PTPN11 p.E76K and CBL p.Y371H iPSC to discover therapeutically exploitable differences in signal pathway profiles.127 Other investigators demonstrated that iPSC generation was also possible using fibroblasts from patients with NS/MPD and PTPN11 p.D61H or p.G503R mutations.128
Outlook and strategies of therapy
For most patients with JMML, early allogeneic HSCT is the therapy of choice.65,82,85,129 None of the current approaches to therapy prior to HSCT (summarized in Loh130 ) has been shown to reduce relapse rate occurring with a cumulative incidence of 35%.82 Targeting downstream effectors of activated Ras is a rational therapeutic approach taken up by the Children’s Oncology Group in a current study with the oral MEK inhibitor trametinib (NCT 03190915) in relapsed JMML. Intrigued by the distinct methylation patterns in JMML and some exceptional responses to hypomethylating agents,92,93 the EWOG-MDS chose a different approach investigating safety and efficacy of azacitidine in children with newly diagnosed JMML (EudraCT Number 2014-002388-13). With the rarity of JMML and the recognition of the distinct biology of its genetic subgroups, an international consensus on a road map for the development of translational research and specifically for future cooperative clinical studies will be of utmost importance. The cross-continental development of a common molecular classifier allowing prospective assignment of DNA methylation categories131 is an important step on this road, and a prerequisite for molecularly driven risk stratification. Exploitation of iPSC technology and currently available xenograft model systems can be expected to provide important insight into which pathways to target and how to combine different therapeutic principles. JMML moved from a rare disease to a group of ultrarare distinct entities. Allowing for mechanisms to keep the group together while ensuring best possible care for every child affected by the disorder will be the clinical challenge for tomorrow.
Authorship
Contribution: C.M.N. and C.F. devised and wrote the manuscript.
Conflict-of-interest disclosure: C.M.N. has a consultancy with Celgene. C.F. declares no competing financial interests.
Correspondence: Charlotte M. Niemeyer, Division of Pediatric Hematology and Oncology, Department of Pediatrics and Adolescent Medicine, Medical Center, Faculty of Medicine, University of Freiburg, Freiburg 79106, Germany; e-mail: charlotte.niemeyer@uniklinik-freiburg.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal