

Abstract
The heterogeneous nature of myelodysplastic syndromes (MDS) demands a complex and personalized variety of therapeutic approaches. Among them, allogeneic hematopoietic stem cell transplantation remains the only potentially curative option and is accessible to only a small number of fit patients. For the majority of patients with MDS, treatment strategies are nonintensive and risk-adapted (by the revised version of the International Prognostic Scoring System), ranging from iron chelation and growth factors to lenalidomide and hypomethylating agents. These approaches are noncurative and aimed instead at improving cytopenias and quality of life and delaying disease progression. These limitations underpin the need for more translational research-based clinical trials in well-defined subgroups of patients with MDS. Indeed, much progress has been made over the past decade in understanding the complex molecular mechanisms underlying MDS. Unfortunately, this has not yet translated into approval of novel treatment options. There is a particularly urgent medical need in patients failing current first-line therapies, such as with erythropoiesis-stimulating or hypomethylating agents. Nevertheless, actual developments are expected to pave the way for exciting novel therapeutic opportunities. This review provides an overview of the current therapeutic landscape in MDS focusing on recent advances in clinical and translational research.
Introduction
Myelodysplastic syndromes (MDS) mainly affect the elderly population, meaning that the majority of patients cannot tolerate intensive therapeutic approaches such as allogeneic hematopoietic stem cell transplantation (allo-HSCT). Treatment is risk-adapted, involving the definition of different goals of therapy (Figure 1) according to the risk status of the patient.1 Over the past 2 decades, only a handful of drugs gained market authorization (Figure 2). Among those few drugs, erythropoiesis-stimulating agents (ESAs) were only recently approved in the European Union (EU) following completion of a placebo-controlled study, despite their use in MDS for >2 decades.2
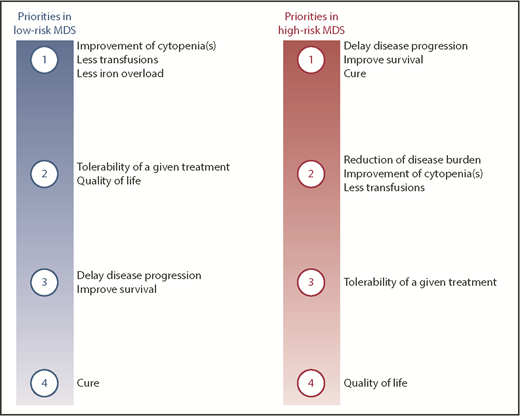
Priorities of therapeutic interventions in patients with MDS according to disease stage. Ranking of potential goals of care in patients with either low- or high-risk MDS.
Priorities of therapeutic interventions in patients with MDS according to disease stage. Ranking of potential goals of care in patients with either low- or high-risk MDS.
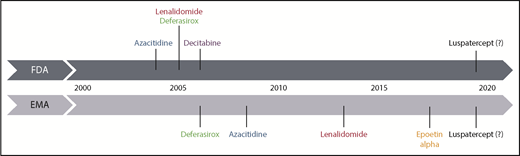
Historical time scale of registration of therapeutic agents for MDS in the EU and United States. Luspatercept is shown as a potential novel drug, which is hoped to get approved in low-risk MDS in the near future. EMA, European Medicines Agency; FDA, US Food and Drug Administration.
Historical time scale of registration of therapeutic agents for MDS in the EU and United States. Luspatercept is shown as a potential novel drug, which is hoped to get approved in low-risk MDS in the near future. EMA, European Medicines Agency; FDA, US Food and Drug Administration.
MDS have a complex pathophysiology, offering multiple points of potential therapeutic intervention. The scope of this review, describing the current therapeutic landscape in MDS, is to highlight the new frontiers that have recently been opened up through a better understanding of the molecular pathophysiology and through clinical and translational research.
Diagnostics in MDS: the first step toward an individual prognostication and treatment
A comprehensive diagnostic workup, nowadays including a panel of molecular abnormalities,3 is mandatory and can provide important prognostic (detailed in scoring systems) and predictive factors for a subsequent response to a given therapy, as is the case for del(5q) and lenalidomide.4
For many years, patients have been classified according to the International Prognostic Scoring System (IPSS) risk score; ie, into “low-risk” MDS (LR-MDS; low/intermediate-1) and “high-risk” MDS (HR-MDS; int-2/high). The revised version of the IPSS (IPSS-R) further stratifies patients into 5 risk groups (LR-MDS being subdivided into IPSS-R very low, low, and intermediate up to 3.5 points5 ), with different outcomes in terms of acute myeloid leukemia (AML) evolution and survival. Nevertheless, the drugs currently available and licensed have all been developed based on the conventional IPSS.
Recent developments in molecular technologies have improved our understanding of the pathogenesis of MDS by identifying somatic mutations in almost every MDS patient.3,6 The identification of these mutations often provides confirmation of clonal disease, being of help for the diagnosis of difficult MDS cases. However, this requires careful consideration because some of the mutations (eg, DNMT3A, TET2) concerned are also present at a lower variant allele frequency in healthy elderly individuals.7 The results of these molecular analyses are meaningful for patients with unexplained cytopenias and suspected myeloid neoplasm. Spliceosome gene mutations and comutations involving TET2, DNMT3A, or ASXL1 are highly predictive for disease evolution in these patients.8 In proven MDS cases, the number and the type of mutations6,9 also have an effect on prognosis: although this is mostly negative, there are exceptions, with SF3B1 mutation conferring a better outcome in patients with no excess of blasts.9 Detailed molecular diagnostics are also crucial for younger patients of intermediate-1 prognosis by IPSS, in whom the presence of 1 or several unfavorable mutations (TP53, ASXL1, RUNX1, EZH2, ETV6) may indicate a more intensive surveillance strategy or even up-front treatment of putative HR-MDS, extending to allo-HSCT.
Current standard of care in treating patients with MDS
In the following sections, current risk-based therapeutic approaches are discussed according to the stage of disease (LR-MDS, HR-MDS) and the type of cytopenia.
LR-MDS
In the case of LR-MDS (IPSS low/intermediate-1, IPSS-R very low, low, intermediate up to 3.5 points5 ), therapy is mainly aimed at improving cytopenia(s) to prevent complications, such as bleeding and severe infections, decreasing transfusion burden, and improving quality of life (Figure 1). In practice, all treatment decisions have to take account of a potential drug-induced deterioration of the patient’s clinical status.
Watchful observation
In a substantial number of patients with LR-MDS, it is inappropriate to go beyond supportive care, because of the mild and asymptomatic level of cytopenia. This can also be a sensible option for patients in other MDS stages who have a low life expectancy because of major comorbidities. Fit LR-MDS patients with an oligosymptomatic cytopenia, no excess of blasts, or poor-risk cytogenetics/molecular profile6 often do not require any treatment other than a regular follow-up.
On the contrary, an intensified surveillance strategy is required for asymptomatic LR-MDS patients with a “higher risk” prognostic profile based on genotype (eg, presence of ASXL1 mutation).6,9 The most important indicators here are the worsening of cytopenia, an increasing number of circulating or bone marrow blasts, and signs of cytogenetic or molecular evolution. In the future, a change of the current observational strategy in this subgroup of patients may be called for when novel and safe treatments capable of modifying the natural history of “very early MDS” disease become available.
Treatment of anemia
Targeting anemia with ESAs
Anemia-related symptoms, such as fatigue, are the most commonly reported symptoms in MDS and the majority of patients require regular red blood cell (RBC) transfusions. This results in iron overload and requires substantial human as well as financial resources (see “Supportive care using iron chelation”).10 Treatment with ESAs (ie, recombinant erythropoietin [EPO] or darbepoetin [DAR]) as single agents is the standard of care in patients with anemia or low transfusion burden. Despite the wide use of ESAs, prospective clinical trials with these drugs have only recently been performed in the EU, leading to the approval of EPO-α, but not DAR.2,11 The reason for the lower response rate with DAR compared with EPO-α in the phase 3 trial (14.7% vs 31.8%) was the use of a less effective 3-week interval of DAR compared with more effective DAR regimens (eg, 500 μg every 2 weeks).2,11 ESAs are considered a first-line treatment of LR-MDS patients displaying pretreatment variables predictive of response to treatment.12 The most decisive of these include a low (<500 IU/L) endogenous EPO level and a transfusion burden <4 U within an 8-week period, according either to the Nordic score12 or to recently refined models.13 Approval of EPO-α in the EU, however, is based on an EPO level <200 IU/L (patients above that threshold did not show response within the registration trial).14 The actual patient selection (Figure 3) allows the subsequent response rate to be easily predicted, avoiding unnecessary patient treatment. Approximately 80% of eligible LR-MDS patients have EPO levels <200 IU/L, whereas only ∼10% exceed 500 IU/L.15 Most responses to ESAs occur within 3 months of treatment and have a median duration of about 15 to 18 months.15 Other predictive factors of response to ESAs include IPSS-R16 and immunophenotype of myeloid cells.17 In eligible patients who either never had or lost response to single-agent ESA, the addition of granulocyte colony-stimulating factor (1-2 μg/kg subcutaneously weekly) may rescue response in up to 20% of cases.18 Granulocyte colony-stimulating factor treatment is of particular benefit to patients with ring sideroblasts (RSs), who may display a shorter duration of response to ESAs than non-RS patients.15
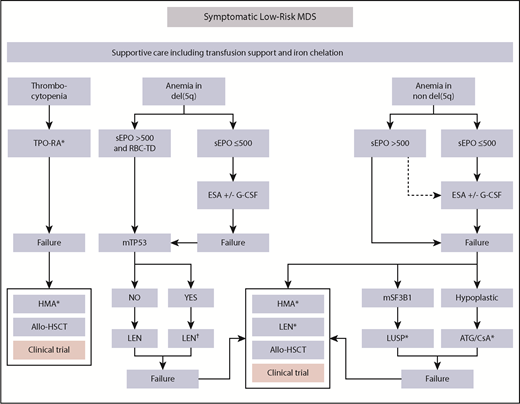
Therapeutic algorithm in LR-MDS patients. Therapeutic options for symptomatic low-risk MDS patients showing anemia or thrombocytopenia. Approximately 80% of eligible patients have EPO levels <200 IU/L, whereas only ∼10% have levels >500 IU/L.15 As a result, ∼90% of LR-MDS patients with anemia are eligible to receive ESAs according to current guidelines. Only 10% to 20% of patients with EPO levels >500 IU/L are unlikely to respond to ESAs and should therefore receive alternative approaches. Notably, prescribing information of the only approved ESA (Eprex) in the EU and United States requires EPO levels <200 IU/L before treatment. At all stages, the patient should be evaluated for a potential clinical trial option. Luspatercept may become a second-line option in the near future in RS+ patients. Thrombopoiesis-stimulating agents are a potential first-line option in patients with clinically meaningful thrombocytopenia. In the presence of TP53-mutation in del(5q), MDS patients should be followed an intensified disease surveillance strategy to detect early signs of disease progression. Dotted arrows indicate potential option in the absence of any therapeutic alternatives. G-CSF, granulocyte colony-stimulating factor; ATG, antithymocyte globulin; CSA, cyclosporine; HMA, hypomethylating agent; LEN, lenalidomide; LUSP, luspatercept; sEPO, serum EPO; TPO-RA, thrombopoietin receptor agonist. *Not presently approved. †Intensified disease surveillance.
Therapeutic algorithm in LR-MDS patients. Therapeutic options for symptomatic low-risk MDS patients showing anemia or thrombocytopenia. Approximately 80% of eligible patients have EPO levels <200 IU/L, whereas only ∼10% have levels >500 IU/L.15 As a result, ∼90% of LR-MDS patients with anemia are eligible to receive ESAs according to current guidelines. Only 10% to 20% of patients with EPO levels >500 IU/L are unlikely to respond to ESAs and should therefore receive alternative approaches. Notably, prescribing information of the only approved ESA (Eprex) in the EU and United States requires EPO levels <200 IU/L before treatment. At all stages, the patient should be evaluated for a potential clinical trial option. Luspatercept may become a second-line option in the near future in RS+ patients. Thrombopoiesis-stimulating agents are a potential first-line option in patients with clinically meaningful thrombocytopenia. In the presence of TP53-mutation in del(5q), MDS patients should be followed an intensified disease surveillance strategy to detect early signs of disease progression. Dotted arrows indicate potential option in the absence of any therapeutic alternatives. G-CSF, granulocyte colony-stimulating factor; ATG, antithymocyte globulin; CSA, cyclosporine; HMA, hypomethylating agent; LEN, lenalidomide; LUSP, luspatercept; sEPO, serum EPO; TPO-RA, thrombopoietin receptor agonist. *Not presently approved. †Intensified disease surveillance.
Targeting anemia in genetically defined del(5q) MDS
ESAs are also a first-line option in del(5q) LR-MDS patients with symptomatic anemia or low transfusion burden and a high likelihood of response (Figure 3).12 However, many of these patients already have excessive EPO levels, which also correlate with a higher clonality rate in progenitor cells17 and predict a lack of or short-lived response to ESAs. For these patients, who also require continuous transfusion support, lenalidomide is the treatment of choice and results in erythroid responses in ∼70% of patients.19-21 The median response duration is 2 years. Approximately 40% of the patients in the MDS 004 study21 had progressed to AML at 5 years.
Interestingly, patients with a TP53 mutation (up to 20% of all patients)22 are less likely to achieve complete cytogenetic remission with lenalidomide and have a higher intrinsic rate of leukemic development compared with patients without a TP53 mutation (5-year cumulative incidences 77% and 24%, respectively23 ). For this reason, patients with a diagnosis of del(5q) LR-MDS harboring or developing a TP53 mutation (during treatment with lenalidomide) should be considered a distinct entity requiring intensified disease surveillance, including regular bone marrow assessment of clonal evolution. Results of the German Lenalidomide Monotherapy in del(5q) (LEMON5) study22 showed that patients with a TP53 mutation display overall lower response rates (RBC transfusion independence [RBC-TI]: 50% vs 75%) and survival. The size of the clone also changes over time in some patients, featuring novel mutations during treatment but without an effect on outcome. Another study24 suggested that MDS patients with a lower TP53 mutation allelic burden (<20%) display a better outcome. Other than TP53, the presence of mutated TET2 and RUNX1 also carries an increased probability of disease progression in del(5q) LR-MDS.25 Patients failing lenalidomide even in the absence of disease progression should be evaluated for allo-HSCT or a potential clinical trial option, whereas hypomethylating agents (HMAs) may be considered in the absence of any trial opportunity (Figure 3).
Second-line treatment of anemia in non-del(5q) MDS
Treatment with ESAs can induce erythroid responses in a meaningful proportion of patients with LR-MDS. Nevertheless, response is mostly transient, suggesting an unmet medical need for novel therapeutic options for patients with anemia or red blood cell transfusion dependency (RBC-TD) who lack or lose a response to ESAs.
Supportive care using iron chelation
Many patients with MDS depend on regular RBC transfusions as part of their supportive care regimen. Consequently, they accumulate excessive amounts of iron (250 mg per RBC unit), which may lead to oversaturation of the physiological systemic iron carrier transferrin and the occurrence of nontransferrin-bound iron together with its reactive fraction, labile plasma iron. Indeed, iron may exert multiple effects that contribute to the pathogenesis and complications of MDS.26 The recently accomplished Myelodysplastic Syndromes (MDS) Event Free Survival With Iron Chelation Therapy trial is the first placebo-controlled study to examine iron chelation in LR-MDS. Available results showed a 36.4% risk reduction in event-free survival (defined by nonfatal events such as worsening cardiac function, hospitalization for congestive heart failure, liver function impairment, liver cirrhosis, transformation to AML) in chelated patients.27 Overall survival data do not show a significant difference, although the data are hardly mature.
The majority of international guidelines recommend a ferritin-guided (at least >1000 ng/mL) chelating approach for the treatment of iron overload in LR-MDS patients.1 The most commonly used iron-chelating drug is deferasirox, which in its new formulation offers an improved tolerability compared with dispersible tablets.28 Chelation should be regarded as mandatory in patients who are scheduled for allo-HSCT (Figure 4 and the following section).
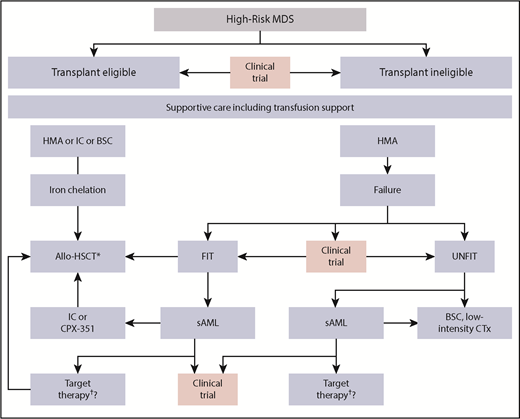
Therapeutic algorithm in HR-MDS patients. The different therapeutic options for HR-MDS patients. The first step is to determine whether the patient is potentially fit and eligible for an allo-HSCT. Pretransplant options may vary depending on disease burden and donor availability, whereas iron chelation is recommended until allo-HSCT. At all stages, the patient should be evaluated for a potential clinical trial option. Patients progressing to AML may take advantage of recent approval of targeted therapies including IDH or FLT3-inhibitors. CTx, chemotherapy; IC, induction chemotherapy; SC, best supportive care; TKI; tyrosine kinase inhibitor. *These could be IDH or FLT3-inhibitors (not presently approved). †Consider posttransplant disease surveillance strategies.
Therapeutic algorithm in HR-MDS patients. The different therapeutic options for HR-MDS patients. The first step is to determine whether the patient is potentially fit and eligible for an allo-HSCT. Pretransplant options may vary depending on disease burden and donor availability, whereas iron chelation is recommended until allo-HSCT. At all stages, the patient should be evaluated for a potential clinical trial option. Patients progressing to AML may take advantage of recent approval of targeted therapies including IDH or FLT3-inhibitors. CTx, chemotherapy; IC, induction chemotherapy; SC, best supportive care; TKI; tyrosine kinase inhibitor. *These could be IDH or FLT3-inhibitors (not presently approved). †Consider posttransplant disease surveillance strategies.
Lenalidomide
A phase 3 placebo-controlled study investigated lenalidomide in non-del(5q) patients with RBC-TD refractory to ESAs.29 Overall, 27% of lenalidomide-treated patients achieved RBC-TI ≥8 weeks, with a median duration of response of 8.2 months. Recently, a randomized trial showed that the combination of lenalidomide and ESA significantly improved erythroid response rate (23.1% vs 39.4%), but not response duration in ESA-resistant LR-MDS patients compared with lenalidomide alone.30 Nevertheless, despite some beneficial clinical effect, lenalidomide will not be registered in this setting because the manufacturer has retracted regulatory submission (Figure 3).
Immunosuppressive agents
Profound immune dysregulation is an increasingly recognized feature of MDS, contributing to ineffective hematopoiesis and driving disease progression.31 Immunosuppressive therapy with antithymocyte globulin (ATG, either horse or rabbit), with or without addition of cyclosporine (CSA), has been investigated in a number of clinical trials, showing trilineage response rates ranging from 16% to 67%.32 Various predictors of response have been described, most notably MDS-single lineage dysplasia (formerly refractory anemia) with absence of RSs, a hypoplastic bone marrow, DR15 HLA type, younger age (<60 years), female sex, trisomy 8, and short duration of transfusion dependence. Interestingly, a large retrospective analysis demonstrated higher responses with horse compared with rabbit ATG and, although the overall response rate was 45%, the presence of SF3B1 mutations negatively affected response.33
Currently, ATG/CSA (horse ATG preferred) is still recommended in clinical routine in rare cases with hypoplastic MDS and normal karyotype failing first-line therapy with growth factors (Figure 3). Very high response rates of 72% were also reported using the anti-CD52 antibody alemtuzumab in a cohort of LR-MDS patients with a high likelihood of response based on HLA-DR15 expression, age, and duration of transfusion dependence.34 Nevertheless, the clinical implementation of this strategy is limited by the drug not being developed further for applications in hematology.
Given the activation of the innate immune system with sustained inflammation in the bone marrow microenvironment35 mediated by MDSC36 and through Toll-like receptor signaling, clinical trials have been recently initiated to target these components by specific antibodies against CD33 (MDSC) or TLR2 (NCT02240706, NCT02363491).
HMAs
Standard or dose-reduced HMAs also have some activity in LR-MDS. Although their use is quite common and covered by US Food and Drug Administration labeling, their use in other regions of the world, including the EU, is rare, because activity has been reported to be very limited after failure to first-line treatment with ESA.37,38 In a French study, RBC-TI was achieved in 16.3% of patients only and could not be improved by the addition of ESA.38 First-line low-dose HMA treatment39 with 3 days of decitabine (DAC) resulted in a 32% RBC-TI rate. Notably, the majority of patients were ESA-naive. Further studies are ongoing (NCT02269280). The oral formulation of azacitidine (AZA; CC-486) provides a more convenient way of administration, allowing the drug to be delivered at lower doses over a prolonged period. However, a phase 3 placebo-controlled study in intermediate-1 risk MDS patients with RBC-TD and thrombocytopenia (NCT01566695) has recently been prematurely terminated because of toxicity.40
Allo-HSCT
The earlier the transplantation is carried out in the disease course, the better are the long-term results. On the other hand, patients with less advanced disease should not be exposed to the substantial risk of mortality of this procedure because of the favorable prognosis with standard treatment alone.41 A recent study by the European Society for Blood and Marrow Transplantation in 246 IPSS low/intermediate-1 patients demonstrated 3-year survival rates of 58% accompanied by a 30% overall nonrelapse mortality rate.42 As a result, patients with LR-MDS failing first-line options and harboring poor-risk features including life-threatening infections, severe thrombocytopenia, RBC-TD, or certain poor-risk molecular abnormalities6 should be considered on a case-by-case basis as appropriate candidates if no clinical trial is available (Figure 3).
Novel approaches to treat anemia in LR-MDS
Erythropoiesis-maturating agents (EMAs)
Both luspatercept and sotatercept are specific activin receptor fusion proteins that act as ligand traps to neutralize negative regulators of late-stage erythropoiesis.43 Luspatercept (ACE-536) has recently shown promising ability to increase hemoglobin with limited toxicity in a phase 2 study in LR-MDS patients.44 In fact, 63% of patients showed erythroid responses, with 38% achieving transfusion independence. Rather than the otherwise familiar predictors of response (such as transfusion burden and EPO levels), it is the presence of RSs or SF3B1 mutations that appeared in this case to define a subgroup with a better response. These findings resulted in a placebo-controlled randomized study of luspatercept in LR-MDS patients with RS-MDS or SF3B1 mutation, refractory to or not eligible for ESA (NCT02631070, A Study of Luspatercept (ACE-536) to Treat Anemia Due to Very Low, Low, or Intermediate Risk Myelodysplastic Syndromes trial). The study met its end point (RBC-TI ≥8 weeks: 37.9% vs 13.2%, P < .0001) with a median duration of response of 30 weeks. It is therefore to be hoped that the data45 will lead to EMA registration of this large subset of MDS patients (Figures 2 and 3).
Sotatercept (ACE-011) differs slightly from luspatercept in respect to ligand affinities, actually having a higher affinity to activin A. However, clinical results with the 2 drugs in LR-MDS are comparable46 and sotatercept will not be further developed in MDS (NCT01999582, NCT01562405).
Hypoxia-inducible factor or telomerase modulation
Roxadustat (FG-4592) is an orally administered hypoxia-inducible factor prolyl hydroxylase inhibitor currently in clinical development for anemia in MDS and chronic kidney disease. Roxadustat promotes erythropoiesis by increasing endogenous EPO levels and improves iron regulation by modulating hepcidin levels. Phase 1 or 2 data in MDS are not yet available, but roxadustat administration in preclinical rodent models has been shown to increase hemoglobin levels. Currently, roxadustat is being assessed in a phase 3, randomized, double-blind, placebo-controlled study for the treatment of anemia in patients with LR-MDS and low RBC transfusion burden (NCT03263091) as well as chronic kidney disease.
Imetelstat is a telomerase inhibitor targeting cells with short telomere lengths and hyperactive telomerase. Telomeres and telomerases are thought to be critical in maintaining normal hematopoiesis. Consecutive shortening of telomeres is thought to lead to reduction of mitotic capacity and ultimately lead to apoptosis. Telomerase hyperactivity can extend this natural limit of mitotic divisions and may promote disease evolution. It has been shown that MDS patient cells have significantly shorter telomeres than those of healthy controls and that high telomerase activity is linked to disease risk.47 Currently, a phase 2/3 study with imetelstat is ongoing in RBC transfusion-dependent and ESA-relapsed or refractory LR-MDS patients. Preliminary results are encouraging, demonstrating that 37% of these patients achieved RBC-TI including reduction of clonal burden.48
Treatment of thrombocytopenia
Thrombocytopenia is present in about one-half of patients with LR-MDS, although severe thrombocytopenia is much less prevalent. Apart from disease-modifying therapies such as HMAs,39 platelet transfusions and TPO-receptor agonists (TPO-RA) are currently the only reasonable treatment options. Romiplostim has been studied in a double-blind, randomized, placebo-controlled clinical trial.49 Thirty-six percent of patients experienced platelet response, associated with a survival benefit compared with nonresponding patients.50 Some patients even demonstrated trilineage responses. The results of a randomized trial in LR-MDS patients treated with eltrombopag, a small molecule TPO-RA, showed 47% platelet responses.51 AML transformation rate was similar between romiplostim and placebo as well as between eltrombopag and placebo in these trials. However, transient elevation of peripheral blasts has been observed in ∼10% of patients and should be carefully monitored. Therefore, although these agents are not formally licensed for MDS, both can be safely used in patients, preferentially within registries or trials and in those patients with no excess of blasts (<5%). Further studies are ongoing (NCT02335268).
HR-MDS
The principal aim of treatment in HR-MDS is to modify the natural course of disease, limiting disease progression, and improving survival rates (Figure 1). Before starting any treatment, the eligibility of the patient for allo-HSCT should be evaluated41 (Figure 3), because this will have an important effect on subsequent workup and patient surveillance.
First-line HMAs
For high-risk patients with MDS who are not suitable for intensive treatment approaches, HMAs (AZA and DAC) represent the only approved therapeutics and current standard or care. There are no prospective data comparing DAC and AZA head-to-head, although retrospective analyses suggest a comparable response activity in MDS.52 Although both drugs are approved in the United States irrespective of disease risk, only AZA is approved for HR-MDS in Europe. The registration trial showed a significant survival benefit (24.5 vs 15 months) of AZA compared with standard of care including intensive chemotherapy.53 The latter is, therefore, only potentially indicated in HR-MDS patients intended to undergo subsequent allo-HSCT because the rate of complete remissions is higher (36% vs 17% in the AZA001 trial53 ) but responses are not durable (see the following section).
Because HMA responses are limited (complete response/partial response/hematologic improvement: 40% to 50%) and mostly short-lived, the identification of factors predicting response is critical. However, the efforts made in this direction have so far met with limited success. Even the role of molecular markers involved in epigenetic regulation, such as TET2 and DNMT3a mutation, is still considered controversial.54-56 Prognostic clinical markers translating into a worse survival (but not response) include presence of peripheral blasts, poor performance status, high transfusion burden, and poor-risk cytogenetics.57 In fact, HMAs are equally active in patients across all cytogenetic subgroups, although responses are especially short-lived in patients with poor-risk abnormalities compared with those with normal karyotype. Overall, the median response duration amounts to ∼1 year, with only a few patients achieving long-lasting remissions. Therefore, there is a tremendous unmet medical need to improve outcome with first-line HMA-based therapies.
Allo-HSCT: indication and timing
Considering the potential treatment-related complications associated with allo-HSCT in MDS patients, the stringent selection process of patients is inevitable; therefore, identification of patient and disease-related factors that predict outcome after allo-HSCT is of the outmost importance.41,42,58 According to a decision model,59 patients with intermediate-2 or HR-MDS by IPSS criteria should be considered for allo-HSCT at the time of diagnosis. Further analyses in older patients receiving reduced-intensity conditioning (RIC) validated these findings also when based on the IPSS-R.41 Allo-HSCT should be considered in patients who have MDS IPSS stage intermediate-2/high or IPSS-R high/very high, are up to the age of 70 to 75 years, in good clinical condition, and have no severe comorbidities.60 Pretransplant comorbidities need to be taken into consideration using the HSCT-specific comorbidity index, because of their potential effect on patient outcome.61
Overall, choosing the optimal moment and integrating allo-HSCT into the therapeutic algorithm remains a challenge in many cases. Although the IPSS-R system was developed mainly to determine the prognostic risk in newly diagnosed MDS patients, its value in predicting posttransplantation outcome has been confirmed in several studies. Although already implemented in the scoring systems, karyotype abnormalities alone represent a significant risk factor for relapse, because certain molecular abnormalities (mainly mutations) in TP53 do.62,63 For these patients, there is a pressing need for posttransplant strategies that prevent or delay relapse. Oral AZA is currently under investigation as maintenance therapy following allo-HSCT in HR-MDS or AML patients (NCT01757535, NCT01835587). Alternative approaches include preemptive MRD-triggered treatments, as recently shown by our group.64
Should patients be treated with an HMA or chemotherapy before allo-HSCT? Large retrospective analyses have demonstrated that the outcome for patients who had received either treatment was comparable after allo-HSCT.65 This finding highlights some of the pitfalls in assessing differences between treatments in a retrospective fashion where patients are selected for various treatments. In the absence of true randomized trials, the value of prior induction chemotherapy therefore remains unclear. However, this strategy might still be the best option in selected patients in the context of innovative fast-track transplant protocols.58,66 The availability of CPX-351 (Vyxeos, see the following section) may potentially lead to a renaissance of intensive chemotherapy in the future given its higher response rates and better tolerability compared with conventional intensive chemotherapy.67 In the majority of patients eligible for allo-HSCT, HMAs are currently used to at least “bridge” the time up to the identification of a compatible donor.65,68 Notwithstanding these options, there remains a substantial number of patients who display disease progression or severe infectious complications during the first 4 months of therapy and therefore cannot undergo subsequent transplantation.68,69
Because the intensity of transplant conditioning is linked to mortality, the development of RIC regimens has allowed the successful application of allo-HSCT in older patients with MDS. The available evidence to date indicates no survival difference between myeloablative approaches and RIC for patients with MDS based on 2 randomized studies.70,71
It is now widely accepted that systemic iron overload directly contributes to outcome after allo-HSCT in MDS.72-74 The results of the ALLIVE (ALLogeneic Iron inVEstigators) Observational Trial study75 demonstrated that elevated labile plasma iron levels before allo-HSCT predict an increased incidence of infection-related nonrelapse mortality (33% vs 7%) and a decreased overall survival in patients with AML or MDS. Therefore, eligible patients should receive appropriate iron chelation before allo-HSCT.
Novel concepts in the treatment of HR-MDS
Second-generation HMAs
Current efforts in clinical research in this area are aimed largely at improving response rates with single-agent HMA in HR-MDS.76 A more prolonged exposure to HMAs may allow a greater incorporation into DNA, because HMA’s action is S-phase dependent. The new-generation DNA hypomethylating drug guadecitabine is a dinucleotide of DAC and deoxyguanosine, which is resistant to degradation by cytidine deaminase and has a longer half-life and exposure than its active metabolite DAC. The drug is currently in phase 3 clinical trials for elderly patients with MDS failing HMAs. Another oral DAC combination (ASTX727) includes the cytidine deaminase inhibitor cedazuridine, which is being tested in first-line treatment (NCT03306264).
Novel combination partners of HMAs
The combinations of AZA with either lenalidomide, vorinostat, or eltrombopag have recently been investigated in randomized trials and did not show any benefit compared with AZA alone.76,77 A study combining AZA with the histone deacetylase inhibitor pracinostat resulted in conflicting results compared with positive data using the same combination to treat elderly AML patients.78 Pevonedistat, a novel inhibitor of the NEDD8-activating enzyme, has shown synergistic antileukemic activity with AZA in preclinical AML models, which has been confirmed in a recent phase 1b study.79 A randomized, controlled phase 3 study (NCT03268954) is currently comparing this combination with single-agent AZA in HR-MDS and oligoblastic AML. IDH mutations are quite common in MDS (10% to 15% of patients), and single-agent data with either IDH1 or IDH2 inhibitors (enasidenib,80 ivosidenib81 ) in relapsed AML including a small MDS subgroup (overall response rate, 59%80 ) are promising and have resulted in first-line combination studies in MDS (NCT03383575, NCT02719574).
The trial testing the combination of venetoclax with AZA in patients with HR-MDS, including HMA failure, is currently recruiting (NCT02966782). This treatment has shown impressive response rates in elderly patients with AML.82,83 It is also known that HMAs dampen immune responses by upregulating inhibitory immune-checkpoint molecule expression, which may in turn contribute to HMA resistance. Single-agent checkpoint modulation may also have the potential to act as a disease-modifying agent.84 Multiple clinical trials in progress76 are evaluating the combination of HMAs with different immune-checkpoint inhibitors (NCT02397720, NCT02775903, NCT02508870), also as second-line therapy. The jury is still out as to which subset of patients may benefit from this approach.
Targeting TP53
Outcomes for patients with HR-MDS and a TP53 gene mutation are generally poor even after allo-HSCT.63 A 10-day regimen of DAC showed very good efficacy in patients with TP53-mutated MDS or AML, leading to transient clearance of mutant cells.85 Such effects have been similarly but less frequently observed with 5 days of DAC86 and with AZA.87,88 Therefore, in the absence of prospective comparative trials (AZA vs DAC), there are no grounds for prioritizing a specific HMA for patients bearing a TP53 mutation. A novel approach is offered by the modulation of the transcriptional activity of mutant TP53 by drugs such as APR-246.89 Preliminary data in combination with HMAs are looking promising and studies are ongoing (NCT03072043). Another appealing strategy to reactivate TP53-mediated effects in nonmutated cases is to inhibit frequently overexpressed suppressor proteins of TP53 (MDMX, MDM2; eg, by stapled peptide ALRN-6924 [NCT02909972]).
Options in patients failing HMA therapy
Although HMAs are active in roughly one-half of HR-MDS patients, the majority either fail to respond or lose an initial response. The subsequent outcome of these patients is mostly poor, with a median survival of <6 months. So far, no approved drug has demonstrated a survival advantage compared with the current standard of care and, although allo-HSCT remains the only potentially curative option, it is available only to a small subset of medically fit patients (Figure 4). Therefore, there is a tremendous clinical need for novel therapies in this segment (Figure 5).
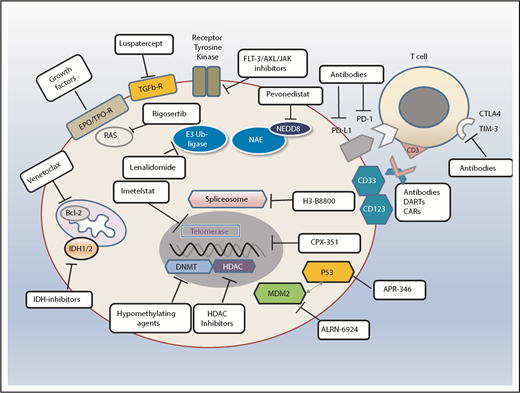
Standards and perspectives of therapeutic options in patients with MDS. Different therapeutic avenues in current clinical practice or ongoing clinical trials. Also shown are examples of different types of agents depending on their specific target or mode of action. NEDD is an ubiquitin-like protein. CAR, chimeric antigen receptor; DART, dual affinity retargeting protein; HDAC, histone deacetylase; NEDD8, neural precursor cell expressed developmentally downregulated protein 8.
Standards and perspectives of therapeutic options in patients with MDS. Different therapeutic avenues in current clinical practice or ongoing clinical trials. Also shown are examples of different types of agents depending on their specific target or mode of action. NEDD is an ubiquitin-like protein. CAR, chimeric antigen receptor; DART, dual affinity retargeting protein; HDAC, histone deacetylase; NEDD8, neural precursor cell expressed developmentally downregulated protein 8.
A switch to currently available HMAs is likely to result in transient responses in a small subset of patients, although an ongoing phase 3 trial with the novel HMA guadecitabine (NCT02907359, see previous section) has shown some encouraging results in an earlier phase 2 study (reviewed in Montalban-Bravo et al90 ).
In clinically fit patients with secondary AML and normal karyotype who fail first-line treatment with HMAs, intensive chemotherapy can be considered if there is a prospect of subsequent allo-HSCT. However, the initial remission rates of 30% to 40% of patients with induction chemotherapy are in fact mostly short-lived.91
CPX-351 (Vyxeos) is a novel liposomal formulation with a fixed 5:1 molar ratio of cytarabine and daunorubicin and has recently received market authorization for the primary treatment of secondary or therapy-related AML.92 Within an open-label phase 3 study, older patients with high-risk AML (including secondary to higher risk MDS and failure to prior HMA) were randomized to receive induction therapy with CPX-351 or “7 + 3.” Median overall survival was 9.56 months with CPX-351 vs 5.95 months with “7 + 3,” which amounts to a significant 31% improvement favoring CPX-351.67 Thus, CPX-351 is a potentially interesting option for fit HR-MDS patients progressing into AML even after HMA treatment who are eligible for intensive treatment including consecutive allo-HSCT. Studies in MDS are also underway.
The first randomized phase 3 study to be completed in the setting of HMA failure compared standard of care (mostly supportive care only) with rigosertib, a multikinase inhibitor. Unfortunately, the primary end point of a demonstrable survival advantage with rigosertib was not met.93 A further subgroup analysis defined that patients with early failure to HMAs (within the first 9 months) seemed to benefit most from rigosertib. Based on this, a consecutive study has recently been launched with rigosertib in this specific subgroup of HR-MDS patients (NCT02562443).
Other potentially available approaches include the use of targeted molecular therapies (eg, with IDH or FLT3-inhibitors).80,81,94 IDH181 or IDH2 inhibitors80 have shown meaningful clinical activity in relapsed AML including a small subset of MDS patients.80 FLT-3 inhibitors have already been approved in the United States for second-line treatment of patients with AML and may thus offer a therapeutic option in rare FLT-3 mutated cases with disease progression (Figure 4). Although those molecular aberrations concerned are rare in MDS compared with AML, many patients have already evolved to secondary AML at the time of failure to HMA, showing secondary clonal evolution and acquisition of such mutations.95
The most common mutations in MDS affect the splicing machinery and the oral spliceosome-modulator H3B-8800, which is currently being assessed in a phase 1 study, and might be a promising targeted drug for this patient cohort.96
The current surge in research and development of immune therapeutics (Figure 5), including chimeric antigen receptor T cells and bispecific antibodies or dual affinity retargeting proteins offers further hope for the future. So far, clinical data in MDS are very limited but underscore the feasibility of such approaches. A phase 1 trial of the CD3/CD123 antibody flotetuzumab has reported some encouraging preliminary results in patients with advanced MDS and AML.97 CD123 is expressed at high levels on leukemic stem cells and is differentially overexpressed in AML and MDS patients. However, the anti-CD123 monoclonal antibody talacotuzumab recently failed to show any meaningful activity in advanced MDS and AML (data not shown, NCT03011034).
Summary and future perspectives
Anemia associated with MDS is routinely treated by ESAs or lenalidomide in LR-MDS with isolated del(5q) and by HMAs in HR-MDS. Thrombocytopenia and neutropenia are less frequent than anemia in LR-MDS, and their treatment has proven more difficult. This paucity of available therapeutic options generates a tremendous medical need for new treatments in MDS (Figure 5). In this situation, it is important to offer available clinical trial options to every patient with MDS across all stages of the disease. Multiple novel therapies are in development or under investigation. Luspatercept is hoped to become clinically available in the near future for a subset of patients with LR-MDS. HR-MDS patients are likely to benefit from the recent clinical research developments in elderly AML patients studying novel HMA combinations, because the 2 diseases are now considered a “biological continuum.” Together with our improved understanding of the molecular and immunological pathophysiology of MDS, it is expected that emerging data will evolve and translate into more and better personalized treatment options for patients with MDS.
Acknowledgments
The author thanks Silke Gloaguen, Lionel Ades, Anne Sophie Kubasch, Francesca Vinchi, Michael Cross, and Ari Giagounidis for constructive discussions during the development of the manuscript.
U.P. received support from Deutsche Jose-Carreras Leukämiestiftung and Boll-Stiftung.
Authorship
Contribution: U.P. designed and wrote the paper.
Conflict-of-interest disclosure: U.P. has received research support and honoraria from Celgene, Amgen, Janssen, and Novartis.
Correspondence: Uwe Platzbecker, Medical Clinic and Policlinic 1, Hematology and Cellular Therapy, Leipzig University Hospital, Leipzig, Johannisallee 32 A, 04103 Leipzig, Germany; e-mail: uwe.platzbecker@medizin.uni-leipzig.de.
