

Key Points
Expression of murine Cebpe is regulated by an enhancer located 6 kb downstream from its transcriptional start site.
Deletion of the +6-kb enhancer in mice leads to a complete block in terminal differentiation of granulocytes.

Abstract
CCAAT/enhancer binding protein ε (CEBPE) is an essential transcription factor for granulocytic differentiation. Mutations of CEBPE occur in individuals with neutrophil-specific granule deficiency (SGD), which is characterized by defects in neutrophil maturation. Cebpe-knockout mice also exhibit defects in terminal differentiation of granulocytes, a phenotype reminiscent of SGD. Analysis of DNase I hypersensitive sites sequencing data revealed an open chromatin region 6 kb downstream of the transcriptional start site of Cebpe in murine myeloid cells. We identified an interaction between this +6-kb region and the core promoter of Cebpe using circular chromosome conformation capture sequencing (4C-seq). To understand the role of this putative enhancer in transcriptional regulation of Cebpe, we targeted it using catalytically inactive Cas9 fused to Krüppel-associated box (KRAB) domain and observed a significant downregulation of transcript and protein levels of CEBPE in cells expressing guide RNA targeting the +6-kb region. To further investigate the role of this novel enhancer further in myelopoiesis, we generated mice with deletion of this region using CRISPR/Cas9 technology. Germline deletion of the +6-kb enhancer resulted in reduced levels of CEBPE and its target genes and caused a severe block in granulocytic differentiation. We also identified binding of CEBPA and CEBPE to the +6-kb enhancer, which suggests their role in regulating the expression of Cebpe. In summary, we have identified a novel enhancer crucial for regulating expression of Cebpe and required for normal granulocytic differentiation.
Introduction
Mature granulocytes arise from hematopoietic stem cells via a cascade of events that involve myeloid lineage commitment, proliferation, and differentiation.1-5 The terminal phases of granulopoiesis are marked by distinct transcriptional changes, cytoplasmic granule formation, and changes in expression of cell surface markers.6-9 This process is regulated by a set of key transcription factors (TFs), including the CCAAT/enhancer binding protein (C/EBP) family proteins, PU.1, and Gfi-1.10-13 We and other investigators have demonstrated that CEBPE regulates transition from the promyelocyte stage to the myelocyte stage of neutrophil development.14
CEBPE is a member of the C/EBP family of TFs involved in myeloid cell development and induction of several inflammatory mediators.15,16 It is expressed in a stage-specific manner during granulopoiesis and is indispensable for secondary and tertiary granule formation.17 The essential role of CEBPE in granulopoiesis is illustrated in Cebpe-knockout (KO) mice, which display a block in terminal differentiation and the absence of secondary granule proteins. Cebpe-KO mice develop normally, except that they fail to produce functional neutrophils and eosinophils. Neutrophils from these mice have impaired chemotaxis and bactericidal activity.18 Germline mutations in the CEBPE gene have been detected in patients with neutrophil-specific granule deficiency. Their neutrophils display atypical bilobed nuclei, lack expression of granule proteins, and these patients suffer from frequent bacterial infections.19-21
In this study, we aimed to identify regulatory elements that control the lineage-restricted expression of CEBPE. Because active gene-regulatory elements are key to understanding transcriptional control governing biological processes like cell-type specificity and differentiation, we focused the study on identification of an enhancer for Cebpe that is critical for granulocytic differentiation. We identified a region +6 kb from the mouse Cebpe transcriptional start site (TSS), which is characterized by open chromatin exclusively in myeloid cells and is essential for the expression of Cebpe. We demonstrated that germline deletion of this novel enhancer in mice resulted in a complete loss of Cebpe transcript and protein levels, accompanied by a severe block in granulocytic differentiation. We provide evidence that CEBPA and CEBPE bind to the +6-kb enhancer to regulate transcription of Cebpe. These findings demonstrate that the +6-kb Cebpe enhancer is pivotal to transcriptional regulation of Cebpe. We also identified an open chromatin region 7 kb downstream of human CEBPE that might be an ortholog of the murine +6-kb enhancer and consists of binding sites for multiple hematopoietic TFs and may serve as a putative enhancer in human cells.
Methods
Chromosome conformation capture-on-chip and sequencing
Circular chromosome conformation capture sequencing (4C-seq) was performed as described previously22 with slight modifications. Experiments were performed on 2 biological replicates of mouse total bone marrow cells from C57BL/6 mice. In brief, 15 million cells were harvested and cross-linked with 1% formaldehyde, and the nuclei were digested using HindIII-HF (New England Biolabs) overnight. Locations of the HindIII sites are provided in supplemental Figure 1, available on the Blood Web site. Following proximity ligation, DNA was reverse cross-linked and purified using phenol/chloroform extraction, and the ligated circular DNA (3C library) was precipitated with ethanol. The chromosome conformation capture library (3C library) was digested with DpnII (New England Biolabs) overnight, followed by proximity ligation and purification to obtain the 4C library. The 4C-seq library was generated by performing nested inverse polymerase chain reaction (PCR) using Phusion DNA polymerase (Thermo Scientific) with the primers listed in supplemental Table 1. DNA was amplified using outer primers in the first PCR, and one tenth of the first PCR product was used as a template in the second PCR using nested primers. 4C-seq libraries were obtained through gel excision of the amplicon on a 4% to 20% gradient TBE Gel (Thermo Fisher Scientific). The libraries were precipitated with ethanol in the presence of GlycoBlue (Thermo Fisher Scientific). The multiplexed 4C-seq libraries were pooled in equal molar ratios, purified using Agencourt AMPure XP (Beckman Coulter) in a 0.8 bead/DNA sample ratio, and sequenced on an MiSeq platform (Illumina) with 2 × 250 bp reads. A minimum of 1 million sequencing reads was produced for each library. The long-range genomic-interaction regions generated by the 4C-seq experiments were analyzed using R package r3CSeq v1.22.0.23 Briefly, raw reads from each replicate were aligned to the masked version of the reference mouse genome (masked for the gap, repetitive, and ambiguous sequences) downloaded from the R Bioconductor repository (BSgenome.Mmusculus.UCSC.mm10.masked). Chromosome 14 was selected as the viewpoint chromosome for Cebpe, with HindIII as its restriction enzyme. A nonoverlapping window size of 5 kb was selected to identify the interacting regions. Numbers of mapped reads for each window were counted and normalized to obtain reads per million per window values to perform the statistical analysis. Interaction regions were plotted based on the significance level.
Cell culture and lentiviral transduction
32D cells were cultured in Iscove modified Dulbecco's medium containing 10% fetal bovine serum, penicillin/streptomycin, and 1 ng/mL recombinant mouse interleukin-3. To obtain 32D cells stably expressing catalytically inactive Cas9 fused to Krüppel-associated box (KRAB) domain (dCas9-KRAB) and single guide RNA (sgRNA), cells were transduced with lentiviral particles by centrifugation at 1000g for 90 minutes in the presence of 8 μg/mL polybrene (Sigma-Aldrich). GFP+ cells (expressing dCas9-KRAB sgRNA) were sorted 3 days after transduction and expanded.
Flow cytometry
Single-cell suspensions were incubated with fluorochrome-conjugated antibodies for 30 minutes on ice. Cells were washed with 2% fetal bovine serum/phosphate-buffered saline and resuspended in SYTOX Blue Dead Cell Stain (Thermo Fisher Scientific). Flow cytometric analysis was performed on a FACS LSR II flow cytometer, and sorting of cells was performed on a FACSAria cell sorter (both from BD Biosciences). Data were analyzed using FACSDiva software (BD Biosciences). The antibodies used in this study are detailed in supplemental Table 1.
RNA isolation, reverse transcription, and qRT-PCR
RNA from cell sorted murine granulocytes and 32D cells was isolated using an RNeasy Micro Kit or an RNeasy Mini Kit (QIAGEN), depending on the number of cells. Complementary DNA was prepared using Maxima Reverse Transcriptase (Thermo Fisher Scientific). Primer sequences used for quantitative reverse transcription PCR (qRT-PCR) are listed in supplemental Table 1.
Immunoblotting
32D cells or sorted immature granulocytes were lysed in 2× gel loading dye, and proteins were resolved on 12% sodium dodecyl sulfate (SDS) polyacrylamide gel. Proteins were transferred to polyvinylidene difluoride membranes and probed with primary antibodies overnight. Membranes were incubated with appropriate horseradish peroxidase–conjugated secondary antibodies for 1 hour and developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Antibodies used are listed in supplemental Table 1.
RNA sequencing and gene-expression analysis
RNA sequencing was done as previously reported.24 In brief, complementary DNA libraries were prepared from poly-A–selected RNA using a TruSeq RNA sample kit (Illumina). Libraries were sequenced on a HiSeq 4000, and 100 bp paired-end reads were aligned to murine reference transcriptome (GRCm38/mm10; Ensemble version 84) using Kallisto (version 0.43.0). Differential analysis was performed using DESeq2. Gene expression was quantified in fragments per kilobase of transcript per million (FPKM) units using DESeq2 FPKM command and was used for all downstream analysis and plotting. All other test statistics and plotting were performed using R 3.4.0. Gene ontology was performed on differentially expressed genes using goseq Bioconductor package (version 1.20.0). Resulting P values were adjusted for false discovery rate. For gene set enrichment analysis, we used all “active transcripts” with mean expression of 0.5 FPKM to identify significantly enriched gene sets among MSigDB C2 gene sets.
ChIP-seq and ChIP-qPCR
DNA–protein complexes were cross-linked with 1% formaldehyde for 10 minutes, followed by neutralization with 0.2 M glycine for 5 minutes. Cells were lysed, and chromatin was sonicated in lysis buffer (1% SDS, 50 mM Tris-HCl, 5 mM EDTA) at 4°C using a Diagenode Bioruptor. Sonicated chromatin was incubated overnight at 4°C with antibodies against CEBPA, CEBPE, or appropriate immunoglobulin G and a 1:1 mixture of Dynabeads Protein A and Protein G. Bead–chromatin complexes were washed, and the chromatin was eluted in 1% SDS and 0.1 M sodium bicarbonate and reverse cross-linked at 65°C for 16 hours. Immunoprecipitated DNA was extracted using a QIAquick PCR Purification Kit (QIAGEN) and quantified using a Qubit Fluorometer (Life Technologies). For chromatin immunoprecipitation sequencing (ChIP-seq), adapter sequences were ligated to DNA fragments, followed by polymerase chain reaction (PCR) amplification and size selection (100-300 bp). Libraries of chromatin immuno-precipitated DNA were sequenced on a HiSeq 4000 (Illumina). For chromatin immunoprecipitation-quantitative PCR (ChIP-qPCR) analysis, input and immunoprecipitated DNA were amplified using 3 primer pairs. Primer sequences used for ChIP-qPCR and antibodies used for pull-down are listed in supplemental Table 1.
Electrophoretic mobility shift assay
293T cells in 100-mm dishes were transfected with 1 μg pCDNA3.1(−) empty vector or Cebpe-expressing vector using jetPRIME transfection reagent (Polypus), according to the manufacturer’s instructions. After transfection, cells were cultured for 48 hours, and nuclear extracts were prepared using NE-PER reagent (Thermo Scientific). Double-stranded oligonucleotide probes were labeled using a 3′ Biotin End DNA Labeling Kit (Thermo Scientific), following the manufacturer’s instructions. The following probes were used: target oligonucleotide 1 (5′-CGGGACGGTTTGCAAAACTCCCAGTAGC-3′), mutant oligonucleotide 1 (5′-CGGGACGGCGCCGCGCCATCCCAGTAGC-3′), target oligonucleotide 2 (5′-CCCAGAGATTGCCTCACTCCCGGGG-3′), and mutant oligonucleotide 2 (5′-CCCAGAGTGGCTTGGCGGCCCGGGG-3′). Binding reaction and detection were carried out using a LightShift Chemiluminescent EMSA Kit (Thermo Scientific). DNA–protein complexes were resolved on native 10% polyacrylamide–TBE gels.
Luciferase reporter assay
The 1240-bp +6-kb enhancer region (chr14: 54 705 845-54 707 084) was amplified as 3 fragments, peak 1 (0-570 bp), peak 2 (571-1240 bp), and both peaks (0-1240 bp), using genomic DNA extracted from murine bone marrow cells and subcloned into pGL4–basic vector (Promega, Madison, WI). 293T cells were transfected with pCDNA-Cebpe along with pGL4–basic vector, pGL4–6kb-peak1, pGL4–6kb-peak2, or pGL4–6kb-peaks1+2 using jetPRIME reagent (Polypus). Renilla basic vector was cotransfected as a control for normalization of luciferase activity. Luciferase activity was measured 24 hours after transfection using a Promega Dual-Glo assay kit, as per the manufacturer’s instructions.
Mice
Cebpe-KO mice have been described previously.18 Mice were maintained on a C57BL/6J genetic background at the animal facility of the Comparative Medicine Centre, National University of Singapore (NUS). +6-kb enhancer deletion (Δ+6 kb enh) mice were generated using CRISPR-Cas9 technology. Two pairs of guide RNAs (ToolGen, Seoul, South Korea) were designed targeting the sequences flanking the +6-kb enhancer region (supplemental Figure 2). Fifteen nanograms of each guide RNA and 30 ng of Cas9 nickase or wildtype Cas9 protein were microinjected in 1-cell–stage zygotes derived from C57BL/6 mice, which were then implanted into foster mice. Heterozygous founder mice were backcrossed with C57BL/6 mice to establish a colony of Δ+6 kb enh mice. Deletion of +6-kb enhancer was verified by PCR and Sanger sequencing of genomic DNA from mouse tails. All mouse experiments were approved by the Institutional Animal Care and Use Committee at NUS.
Statistical analysis
A 2-sided unpaired Student t test was used to determine the statistical significance of the experimental results. Data are shown as mean ± standard deviation. P < .05 is considered statistically significant.
Results
Genomic region 6 kb downstream of CEBPE TSS has open chromatin associated with active DNA regulatory elements in myeloid cells
We sought genomic regions that contribute to lineage-restricted expression of CEBPE. Because enhancers are regulatory elements characterized by increased chromatin accessibility and histone modifications, we analyzed publically available DNase I hypersensitive sites sequencing data sets for open chromatin regions enriched exclusively in myeloid cells (supplemental Table 2). A unique open chromatin region in mouse myeloid cells (416B) (chr14: 54 705 845-54 707 084 was noted +6 kb downstream of the TSS of Cebpe (Figure 1A).25 Further examination of H3K4me1 and H3K27ac (marks of active enhancers) and H3K4me3 (mark of active promoters) histone modifications indicated enriched signals for all 3 marks around the +6-kb region in mature granulocytes compared with other lineages (Figure 1B).26 Analysis of the H3K27ac signature around the +6-kb region clearly revealed higher H3K27ac binding in hematopoietic cells committed to the myeloid lineage compared with nonhematopoietic cells (supplemental Figure 3).27 Because binding and recruitment of P300 are linked to enhancer activity, we also analyzed P300 ChIP-seq data and observed that its binding was enriched at the +6-kb region specifically in myeloid (416B) cells (Figure 1C).28-30 Promoter enhancer interactions are usually confined within topologically associated domains (TADs) and sub-TADs, which are marked by CTCF and cohesin complex binding. We identified that the genomic region encompassing the +6-kb open chromatin region and the Cebpe gene was flanked by CTCF and Rad21 (subunit of cohesin complex) binding in the in vitro–differentiated neutrophils from a murine promyeloid cell line ECOMG (Figure 1D).31 Further analysis of Hi-C data from murine neutrophils also revealed that the region encompassing the Cebpe promoter and the +6-kb region were located within a TAD31 (supplemental Figure 4). All of these observations suggest that this +6-kb region could act as a putative enhancer for Cebpe.
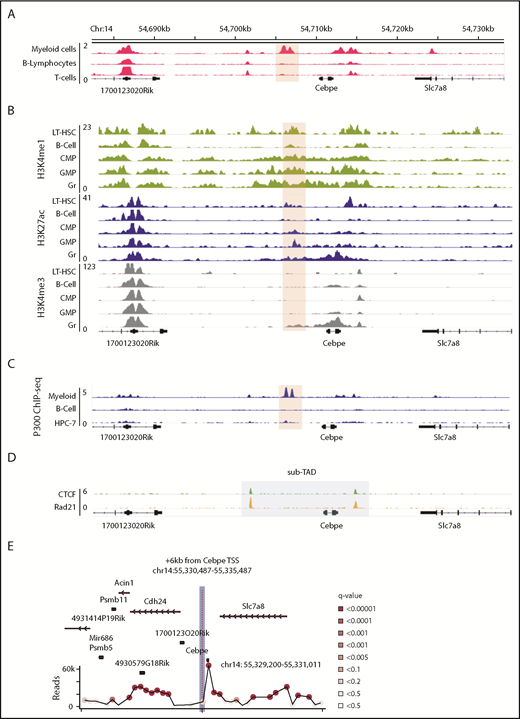
Genomic region 6 kb downstream of Cebpe TSS has properties of an enhancer. (A) Publically available DNase I hypersensitive sites sequencing data for 416B myeloid cells, B lymphocytes, and T cells show that a region +6 kb from Cebpe TSS (region encompassing the +6-kb enhancer has been highlighted) is an open chromatin specifically in myeloid cells.25 (B) Comparison of H3K4me1, H3K27ac, and H3K4me3 ChIP-seq signal in murine long-term hematopoietic stem cells (LT-HSC), B cells, common myeloid progenitors (CMP), granulocyte monocyte progenitors (GMP), and granulocytes (Gr).26 (C) IGV tracks depict P300 ChIP-seq in 416B myeloid cells, B cells, and a hematopoietic progenitor cell line (HPC-7).28-30 (D) +6-kb region is enclosed within a sub-TAD (shaded area), marked by CTCF and Rad21 binding in in vitro–differentiated ECOMG cells.31 (E) 4C-seq profile of murine bone marrow cells reveals strong interaction of the +6-kb enhancer with a region encompassing the Cebpe promoter (chr14: 55 330 487-55 335 487 mm9). Viewpoint region is the shaded area. Circles represent significant interactions detected in our 4C-seq experiments.
Genomic region 6 kb downstream of Cebpe TSS has properties of an enhancer. (A) Publically available DNase I hypersensitive sites sequencing data for 416B myeloid cells, B lymphocytes, and T cells show that a region +6 kb from Cebpe TSS (region encompassing the +6-kb enhancer has been highlighted) is an open chromatin specifically in myeloid cells.25 (B) Comparison of H3K4me1, H3K27ac, and H3K4me3 ChIP-seq signal in murine long-term hematopoietic stem cells (LT-HSC), B cells, common myeloid progenitors (CMP), granulocyte monocyte progenitors (GMP), and granulocytes (Gr).26 (C) IGV tracks depict P300 ChIP-seq in 416B myeloid cells, B cells, and a hematopoietic progenitor cell line (HPC-7).28-30 (D) +6-kb region is enclosed within a sub-TAD (shaded area), marked by CTCF and Rad21 binding in in vitro–differentiated ECOMG cells.31 (E) 4C-seq profile of murine bone marrow cells reveals strong interaction of the +6-kb enhancer with a region encompassing the Cebpe promoter (chr14: 55 330 487-55 335 487 mm9). Viewpoint region is the shaded area. Circles represent significant interactions detected in our 4C-seq experiments.
+6-kb region contacts the core promoter of Cebpe
To verify whether this open chromatin region 6 kb downstream of the Cebpe gene might act as an enhancer to transcriptionally regulate Cebpe, we applied high-resolution 4C-seq in mouse bone marrow cells, using the +6-kb region as the viewpoint. Interaction with a region that encompasses the promoter of Cebpe was highly significant in 2 independent experiments (Figure 1E; supplemental Figure 5; supplemental Table 3). We carried out analyses using window- and fragment-based approaches and observed significant interaction of the enhancer region with the locus containing the Cebpe promoter (supplemental Figure 5). This verified spatial proximity between the Cebpe TSS and the region 6 kb downstream, further supporting our premise that the +6-kb region potentially regulates expression of Cebpe.
+6-kb enhancer regulates Cebpe expression in myeloid cells
To test the transcription regulating potential of the +6-kb region, we used dCas9-KRAB. We designed 6 guide RNAs covering the +6-kb region (sgRNA target sequences are listed in supplemental Table 1) and generated stably transduced 32D cells. Four of the 6 sgRNAs effectively repressed expression of CEBPE (Figure 2A). RNA sequencing of these enhancer-repressed cells revealed downregulation of known CEBPE targets, including lactoferrin (Ltf), neutrophil granule protein (Ngp), and other genes involved in myeloid cell function, S100 calcium binding protein A8 (S100a8) and S100 calcium binding protein A9 (S100a9) (Figure 2B), which was validated using qRT-PCR (Figure 2C). Gene ontology and gene set enrichment analysis of downregulated genes revealed a strong enrichment for pathways implicated in myeloid development and function (supplemental Figure 6). Comparison of gene expression profile of the +6-kb enhancer-repressed 32D cells with immature granulocytes from Cebpe-KO mice also revealed a significant overlap, with ∼95 genes commonly downregulated in the 2 conditions (Figure 2D). Incubation of 32D cells with granulocyte colony-stimulating factor (G-CSF) is known to cause granulocytic differentiation accompanied by induction of Cebpe expression. We observed impaired expression of Cebpe transcripts, as well as its downstream targets, Ltf and Ngp, upon G-CSF induction of these cells expressing sgRNA targeting the +6-kb enhancer region (Figure 2E). These findings demonstrate that the +6-kb enhancer is critical for regulating transcription of Cebpe.
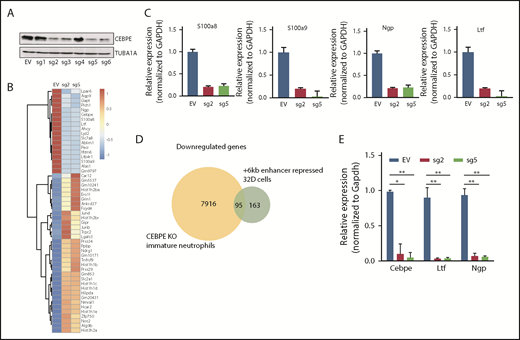
+6-kb enhancer regulates expression of Cebpe in murine myeloid cells. (A) Immunoblot with protein lysates from 32D cells stably transduced with empty vector (EV) or vector expressing dCas9-KRAB and sgRNAs targeting the +6-kb enhancer (sg1-6). Levels of α-tubulin (TUBA1A) were used as loading control. (B) Heat map shows genes that are significantly upregulated or downregulated in 32D cells stably expressing dCas9-KRAB-sg2 (sg2) or dCas9-KRAB-sg5 (sg5) compared with EV. (C) qRT-PCR validation of RNA-sequencing data for selected myeloid-specific genes downregulated in sg2- and sg5-transduced cells. (D) Venn diagram depicts the number of genes commonly downregulated between dCas9-KRAB sg2- and sg5-transduced 32D cells and immature neutrophils from Cebpe-KO mice. (E) qRT-PCR for Cebpe and its target genes (Ltf and Ngp) following G-CSF treatment of EV-, sg2-, and sg5-transduced 32D cells. Y-axis represents relative expression of genes normalized to Gapdh. **P < .01, *P < .05.
+6-kb enhancer regulates expression of Cebpe in murine myeloid cells. (A) Immunoblot with protein lysates from 32D cells stably transduced with empty vector (EV) or vector expressing dCas9-KRAB and sgRNAs targeting the +6-kb enhancer (sg1-6). Levels of α-tubulin (TUBA1A) were used as loading control. (B) Heat map shows genes that are significantly upregulated or downregulated in 32D cells stably expressing dCas9-KRAB-sg2 (sg2) or dCas9-KRAB-sg5 (sg5) compared with EV. (C) qRT-PCR validation of RNA-sequencing data for selected myeloid-specific genes downregulated in sg2- and sg5-transduced cells. (D) Venn diagram depicts the number of genes commonly downregulated between dCas9-KRAB sg2- and sg5-transduced 32D cells and immature neutrophils from Cebpe-KO mice. (E) qRT-PCR for Cebpe and its target genes (Ltf and Ngp) following G-CSF treatment of EV-, sg2-, and sg5-transduced 32D cells. Y-axis represents relative expression of genes normalized to Gapdh. **P < .01, *P < .05.
Deletion of the +6-kb enhancer blocks terminal differentiation of neutrophils
To further assess the consequences of loss of +6-kb enhancer on granulocytic differentiation in vivo, we generated mice with germline deletion of this locus using CRISPR/Cas9 technology (Figure 3A-B). Two independent lines of Δ+6-kb enh mice were established that exhibited identical phenotypes. Flow cytometric analysis of blood and bone marrow cells showed a complete absence of mature granulocytes (CD11b+Gr1hi) in the Δ+6 kb enh mice (Figure 3C-F). This block in granulocytic differentiation is indistinguishable from that observed in Cebpe-KO mice (supplemental Figure 7).
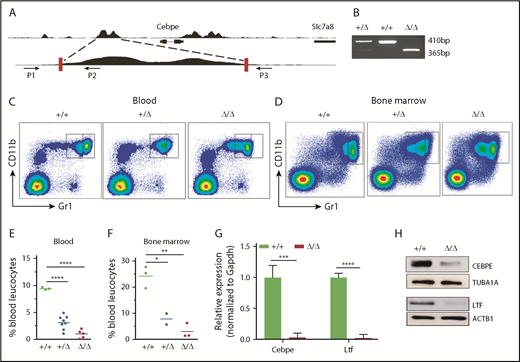
Δ+6 kb enh mice have a complete block in terminal granulopoiesis. (A) Genotyping primers flanking the guide RNA target sequences (red rectangles) generate an amplicon of 410 bp from the WT allele (P1/P2) and an amplicon of 365 bp from the targeted allele (P1/P3). (B) Representative PCR analysis shows genotyping of WT mice (+/+), heterozygous Δ+6 kb enh mice (+/Δ), and homozygous Δ+6 kb enh mice (Δ/Δ). Representative flow cytometric analysis of peripheral blood (C) and bone marrow (D) of WT, heterozygous, and homozygous Δ+6 kb enh mice using granulocyte-specific markers (Gr1 and CD11b). Proportion of mature granulocytes in peripheral blood (E) and bone marrow (F) of WT, heterozygous, and homozygous mice. qRT-PCR (G) and immunoblot (H) for Cebpe and Ltf expression in WT (+/+) and Δ+6 kb enh mice (Δ/Δ). ****P < .0001, ***P < .001, **P < .01, *P < .05.
Δ+6 kb enh mice have a complete block in terminal granulopoiesis. (A) Genotyping primers flanking the guide RNA target sequences (red rectangles) generate an amplicon of 410 bp from the WT allele (P1/P2) and an amplicon of 365 bp from the targeted allele (P1/P3). (B) Representative PCR analysis shows genotyping of WT mice (+/+), heterozygous Δ+6 kb enh mice (+/Δ), and homozygous Δ+6 kb enh mice (Δ/Δ). Representative flow cytometric analysis of peripheral blood (C) and bone marrow (D) of WT, heterozygous, and homozygous Δ+6 kb enh mice using granulocyte-specific markers (Gr1 and CD11b). Proportion of mature granulocytes in peripheral blood (E) and bone marrow (F) of WT, heterozygous, and homozygous mice. qRT-PCR (G) and immunoblot (H) for Cebpe and Ltf expression in WT (+/+) and Δ+6 kb enh mice (Δ/Δ). ****P < .0001, ***P < .001, **P < .01, *P < .05.
+6-kb enhancer controls CEBPE expression in granulocytes
To examine whether deletion of the +6-kb region was accompanied by a decrease in Cebpe expression in vivo, we sorted CD11b+Gr1lo immature granulocytes from bone marrow of wild-type (WT) and Δ+6 kb enh mice. qRT-PCR revealed a marked loss of Cebpe transcript, accompanied by reduced expression of Cebpe target gene Ltf in Δ+6 kb enh mice (Figure 3G). Western blot analysis further verified decreased CEBPE and LTF levels in Δ+6 kb enh mice (Figure 3H).
Multiple TFs essential for hematopoiesis bind to the +6-kb enhancer of Cebpe
To explore TF occupancy at the +6-kb enhancer, we analyzed available ChIP-seq datasets for hematopoiesis-specific factors. Analysis of TF occupancy in a hematopoietic progenitor cell line (HPC7) revealed binding of key TFs, including MEIS1, GFI1B, FLI1, LMO2, LYLI, RUNX1, SPI1, TAL1, and GATA2, to the +6-kb enhancer (Figure 4A).32 Although most of these TFs are involved in early hematopoiesis, they are also expressed in a lineage-restricted manner and are crucial for lineage commitment (supplemental Figure 8). We also noted binding of CEBPA, FLI1, ERG, and SPI1 to the +6-kb enhancer in the granulocyte monocyte progenitor (GMP) population, suggesting its involvement in myeloid differentiation (Figure 4B).28 We focused on CEBPA, a myeloid-specific factor essential for granulopoiesis, whose deficiency leads to a block at the GMP stage in mice.33 We observed that CEBPA binds strongly to the +6-kb enhancer in myeloid precursors in comparison with differentiated myeloid cells, such as macrophages, with almost no binding detected in Lin−Sca+Kit− cells (LSK) cells and B lymphocytes (Figure 4C).27,28,34-36 These findings suggest that CEBPA could be a potential regulator of Cebpe expression; therefore, we analyzed, experimentally, its binding to the +6-kb enhancer.
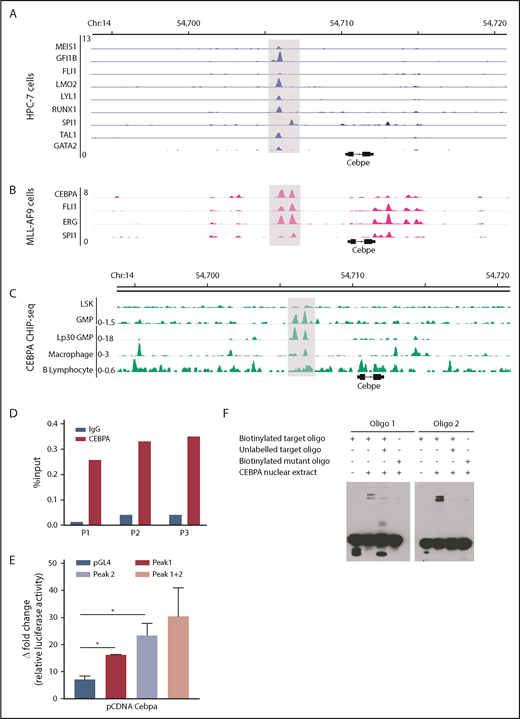
Multiple hematopoietic TFs bind the +6-kb enhancer. (A) ChIP-seq tracks for TFs occupying the +6-kb enhancer region in a hematopoietic progenitor cell line (HPC-7).32 (B) ChIP-seq peaks for CEBPA, FLI1, ERG, and SPI1 at the +6-kb enhancer in sorted GMP cells from mice expressing MLL-AF9 in hematopoietic cells.28 (C) ChIP-seq for CEBPA in different murine hematopoietic cells (region encompassing the +6 kb region has been highlighted). Lp30 GMP, GMPs isolated from mice transplanted with fetal liver from Lp30 mice.34-36 (D) ChIP-qPCR shows enrichment of CEBPA binding within the +6-kb enhancer using 3 primer pairs (P1-P3). (E) Luciferase reporter assay. The +6-kb enhancer sequence was amplified into 3 fragments (peak 1: 0-570 bp, peak 2: 571-1240 bp, and peaks 1+2: 0-1240 bp) and cloned into pGL4–basic vector. Results represent fold induction of relative luciferase activity after normalization to Renilla control in 2 independent experiments, each done in triplicates. (F) EMSA was performed with oligonucleotide (oligo) sequences located within the +6-kb enhancer region. Biotin-labeled target and mutant oligonucleotides were mixed with protein extracts from 293T cells transfected with an empty vector or an expression vector for CEBPA. The reaction mixtures were resolved on native 10% polyacrylamide–TBE gel. Cold competition was carried out with 100-fold molar excess of unlabeled oligo. *P < .05.
Multiple hematopoietic TFs bind the +6-kb enhancer. (A) ChIP-seq tracks for TFs occupying the +6-kb enhancer region in a hematopoietic progenitor cell line (HPC-7).32 (B) ChIP-seq peaks for CEBPA, FLI1, ERG, and SPI1 at the +6-kb enhancer in sorted GMP cells from mice expressing MLL-AF9 in hematopoietic cells.28 (C) ChIP-seq for CEBPA in different murine hematopoietic cells (region encompassing the +6 kb region has been highlighted). Lp30 GMP, GMPs isolated from mice transplanted with fetal liver from Lp30 mice.34-36 (D) ChIP-qPCR shows enrichment of CEBPA binding within the +6-kb enhancer using 3 primer pairs (P1-P3). (E) Luciferase reporter assay. The +6-kb enhancer sequence was amplified into 3 fragments (peak 1: 0-570 bp, peak 2: 571-1240 bp, and peaks 1+2: 0-1240 bp) and cloned into pGL4–basic vector. Results represent fold induction of relative luciferase activity after normalization to Renilla control in 2 independent experiments, each done in triplicates. (F) EMSA was performed with oligonucleotide (oligo) sequences located within the +6-kb enhancer region. Biotin-labeled target and mutant oligonucleotides were mixed with protein extracts from 293T cells transfected with an empty vector or an expression vector for CEBPA. The reaction mixtures were resolved on native 10% polyacrylamide–TBE gel. Cold competition was carried out with 100-fold molar excess of unlabeled oligo. *P < .05.
CEBPA binds the +6-kb enhancer region
To verify binding of CEBPA to the +6-kb enhancer region, we used ChIP-qPCR in murine 32D cells. Significant enrichment of CEBPA binding to the Cebpe enhancer occurred compared with immunoglobulin G control (Figure 4D). To assess whether this binding of CEBPA is functionally relevant, a luciferase reporter assay and an electrophoretic mobility shift assay (EMSA) were performed. For the luciferase reporter assay, we cloned the 1240-bp enhancer sequence as 3 fragments, peak 1 (0-570 bp), peak 2 (571-1240 bp), and both peaks (0-1240 bp), into the pGL4 luciferase vector and cotransfected them with the CEBPA-expression vector into 293T cells. CEBPA markedly transactivated the expression of luciferase reporter in cells transfected with any of the 3 +6-kb enhancer constructs (Figure 4E). Next, biotinylated oligonucleotide probes (located within the +6-kb enhancer) were designed, and EMSAs were performed with the target oligonucleotides and those harboring a mutation in the C/EBP motif. Incubation of biotinylated oligonucleotides with nuclear extract from cells ectopically expressing CEBPA caused a shift in their migration (Figure 4F). This shift decreased markedly when the nuclear extract complex was incubated with a 100-fold molar excess of unlabeled competitor oligonucleotides or when mutant oligonucleotides were used in the experiment (Figure 4F). Taken together, these results substantiate that CEBPA binds the +6-kb region, possibly to regulate Cebpe expression.
CEBPE binds the +6-kb enhancer and may regulate its expression
Our previous ChIP-seq for CEBPE in murine bone marrow cells identified genome-wide chromatin occupancy of CEBPE.37 Interestingly, we also noted binding of CEBPE to its +6-kb enhancer (Figure 5A). A ChIP assay using CEBPE antibody, followed by PCR, validated enriched binding of CEBPE to the +6-kb enhancer (Figure 5B). Luciferase reporter assays also showed an increase in reporter activity upon incubation with CEBPE expression plasmid, although the induction was modest compared with luciferase assay experiments carried out with CEBPA overexpression plasmid (Figures 4E, 5C). In EMSAs, when biotinylated oligonucelotides (located within the +6-kb enhancer) were incubated with nuclear extract from CEBPE overexpressing cells, a shift in migration of the oligonucelotides occurred (Figure 5D). The specificity of this interaction was evidenced by a substantial decrease in the intensity of the protein–oligonucelotide complex in experiments using competitor oligonucelotides (100-fold molar excess) or with oligonucelotides with a mutated sequence in the C/EBP motif (Figure 5D). Taken together, these results imply that CEBPE might autoregulate its expression by binding the +6-kb enhancer. To define this interaction functionally, we stably overexpressed murine CEBPE coding sequence in 32D cells using a lentiviral vector (Figure 5E). We observed significantly higher expression of endogenous CEBPE transcript levels, as indicated by an increase in Cebpe untranslated region (UTR)-specific amplicons in cells stably expressing CEBPE (Figure 5F). This was also accompanied by an induction of CEBPE target genes Ltf and Ngp (Figure 5F). These findings demonstrate that CEBPE binds to the +6-kb enhancer to possibly regulate its own transcription.
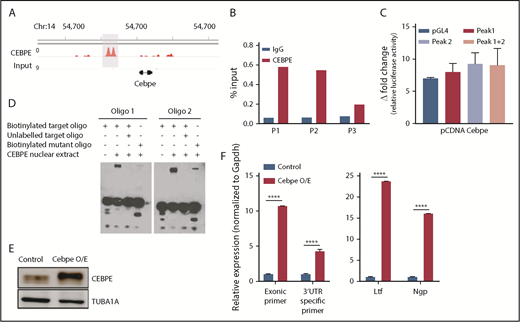
CEBPE binds the +6-kb enhancer and autoregulates its expression. (A) ChIP-seq track depicting occupancy of CEBPE at the +6-kb enhancer in murine bone marrow cells.37 (B) ChIP-qPCR shows enriched binding of CEBPE at the +6-kb enhancer. Three primer pairs were used for PCR analysis. Luciferase reporter assay (C) and EMSA (D) were performed with lysates from CEBPE-expressing cells with target and mutant oligonucleotides (oligos) as described for Figure 4E and 4F. (E) Immunoblot for CEBPE expression in 32D cells stably transduced with lentivirus expressing murine Cebpe (Cebpe O/E). α-Tubulin was used as an endogenous control. (F) qRT-PCR for Cebpe expression in control and Cebpe O/E 32D cells (left panel). Two primer pairs were used: exonic primers that detect total Cebpe transcript levels and 3′-UTR–specific primers that exclusively detect endogenous Cebpe transcripts. qRT-PCR for Ltf and Ngp expression in control and Cebpe O/E 32D cells (right panel). Y-axis represents relative expression of genes normalized to Gapdh. ****P < .0001.
CEBPE binds the +6-kb enhancer and autoregulates its expression. (A) ChIP-seq track depicting occupancy of CEBPE at the +6-kb enhancer in murine bone marrow cells.37 (B) ChIP-qPCR shows enriched binding of CEBPE at the +6-kb enhancer. Three primer pairs were used for PCR analysis. Luciferase reporter assay (C) and EMSA (D) were performed with lysates from CEBPE-expressing cells with target and mutant oligonucleotides (oligos) as described for Figure 4E and 4F. (E) Immunoblot for CEBPE expression in 32D cells stably transduced with lentivirus expressing murine Cebpe (Cebpe O/E). α-Tubulin was used as an endogenous control. (F) qRT-PCR for Cebpe expression in control and Cebpe O/E 32D cells (left panel). Two primer pairs were used: exonic primers that detect total Cebpe transcript levels and 3′-UTR–specific primers that exclusively detect endogenous Cebpe transcripts. qRT-PCR for Ltf and Ngp expression in control and Cebpe O/E 32D cells (right panel). Y-axis represents relative expression of genes normalized to Gapdh. ****P < .0001.
Human CEBPE has a putative enhancer 7 kb downstream from its TSS
Analysis of the murine +6-kb enhancer sequence revealed that this regulatory element is well conserved across several mammalian species (supplemental Figure 9), which prompted us to search for a similar enhancer for CEBPE in humans. Analysis of genomic regions revealed a similar DNase I hypersensitive site +7 kb from CEBPE TSS in NB4 cells (Figure 6A). The region was flanked by CTCF, enriched for binding of P300 and TFs RUNX1, FLI1, and LYL1 in NB4 cells (Figure 6A).27 Analysis of ChIP-seq data for hematopoietic TF binding in Kasumi-1 cells also revealed binding of hematopoietic TFs such as RUNX1, LMO2, SPI1 and CEBPA to the +7-kb region (Figure 6B).38,39 Further, our ChIP-seq experiments for CEBPE in NB4 cells revealed binding of CEBPE to this region (Figure 6C), suggesting autoregulation of CEBPE. These data suggest that this region likely acts as an enhancer for human CEBPE, similar to the +6-kb region that we identified in mouse cells.
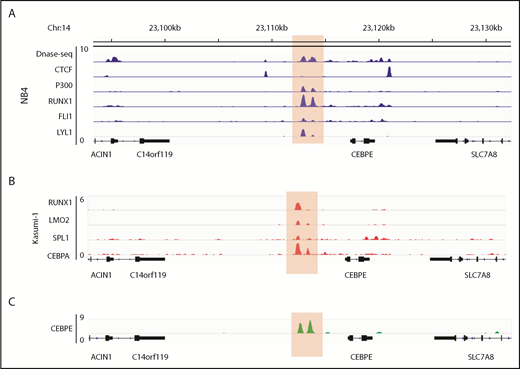
Genomic region 7 kb downstream of human CEBPE TSS is orthologous to the murine +6-kb enhancer. (A) Analysis of publicly available data reveals an open chromatin region 7 kb downstream of CEBPE TSS that is also flanked by CTCF binding and enriched for P300 signal in NB4 cells.27 The region also harbors binding of hematopoietic TFs RUNX1, FLI1, and LYL1 (highlighted). (B) ChIP-seq tracks for TFs occupying the +7-kb region in Kasumi-1 cells (highlighted).38,39 (C) ChIP-seq track depicts occupancy of CEBPE at the +7-kb region in NB4 cells (highlighted).
Genomic region 7 kb downstream of human CEBPE TSS is orthologous to the murine +6-kb enhancer. (A) Analysis of publicly available data reveals an open chromatin region 7 kb downstream of CEBPE TSS that is also flanked by CTCF binding and enriched for P300 signal in NB4 cells.27 The region also harbors binding of hematopoietic TFs RUNX1, FLI1, and LYL1 (highlighted). (B) ChIP-seq tracks for TFs occupying the +7-kb region in Kasumi-1 cells (highlighted).38,39 (C) ChIP-seq track depicts occupancy of CEBPE at the +7-kb region in NB4 cells (highlighted).
Discussion
Enhancers are cis-acting DNA sequences that can increase the transcription of genes through cooperative and synergistic binding of TFs and chromatin-modifying complexes.40 Enhancers function from distal regions in an orientation-independent manner and are characterized by increased chromatin accessibility, distinct patterns of histone modifications and DNA hypomethylation, and bidirectional transcription.41
Using functional genomics and genome-editing approaches, we identified a novel enhancer located 6 kb downstream of Cebpe in murine hematopoietic cells. This genomic locus is located within a TAD flanked by sites of CTCF and cohesin complex binding and exhibits enriched occupancy of P300 and active histone marks, suggesting the importance of this region as a putative enhancer. A defining feature of enhancers is their ability to function as integrated platforms for TF binding, leading to controlled expression of cell-/tissue-specific gene expression.42 Our observation of cell-type–specific TFs (CEBPA and PU.1) binding to the +6-kb enhancer points to a regulatory network that likely controls granulocytic lineage–restricted expression of Cebpe. We demonstrate that deletion of the +6-kb enhancer in mice strikingly reduces CEBPE expression and leads to a complete block in neutrophil differentiation, a phenotype identical to Cebpe-KO mice.
Our preliminary analysis on human leukemic cells points to a similar enhancer in human cells that is 7 kb downstream of CEBPE gene and bound by multiple TFs. We postulate that this genomic locus putatively functions as an enhancer for human CEBPE, similar to our observations with murine cells. Ptasinka et al showed that AML1-ETO binds to the +7-kb region in Kasumi-1 cells, and knockdown of AML1-ETO resulted in an increase in CEBPE expression.38 Moreover, in cells with knockdown of AML1-ETO, an increased occupancy of CEBPA, PU.1, P300, and LMO2 occurred at the +7-kb region.38,39 This suggests that binding of oncogenic factors, such as AML1-ETO, to the region +7 kb from the CEBPE TSS can hinder binding of other cell-type–/lineage-restricted TFs and lead to a block in granulocytic differentiation. Further studies are needed to address whether this +7-kb region in human myeloid cells is essential for expression of CEBPE and regulating granulocytic differentiation, similar to its murine counterpart.
Our experiments also uncover interaction of CEBPE with the novel +6-kb enhancer, suggesting an autoregulatory loop. Inducible vectors expressing CEBPE in cell lines lacking CEBPE could be used to determine the self-regulation of CEBPE or reporter assay experiments, and quantification of 3′-UTR–specific amplicons can be done with overexpression of a DNA-binding mutant of CEBPE. All of these experiments will further our understanding of autoregulation of CEBPE. It would also be highly interesting to investigate whether this enhancer region is altered, either by epigenetic changes or somatic mutations, in SGD patients with no known mutations in CEBPE. We also speculate whether mutations in this +7-kb region may suppress CEBPE expression and ensuing granulopoiesis in acute promyelocytic leukemia, a myeloid malignancy in which we previously detected truncating mutations in CEBPE.43
In summary, we identified a novel enhancer for Cebpe that is indispensable for CEBPE expression and granulocytic differentiation. Our functional assays delineate the enhancer-mediated mechanism of Cebpe regulation, possibly through binding of key TFs to the +6-kb enhancer region. Based on the presence of a similar regulatory region in human cells, we also postulate that this locus may act as an enhancer for CEBPE in human cells.
Presented in abstract form at the 61st annual meeting of the American Society of Hematology, San Diego, CA, 3 December 2018.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the staff of Comparative Medicine, NUS, for support with maintenance of mouse colonies and experiments involving mice; the FACS facility at Cancer Science Institute of Singapore for expert help and support; and the Melamed family and Reuben Yeroushalmi for generous support.
This work was supported by the Leukemia Lymphoma Society of America, the Singapore Ministry of Health’s National Medical Research Council (NMRC) under its Singapore Translational Research (STaR) Investigator Award (NMRC/STaR/0021/2014, H.P.K.), Singapore Ministry of Education Academic Research Fund Tier 2 (MOE2013-T2-2-150), an NMRC Centre Grant awarded to National University Cancer Institute of Singapore (NMRC/CG/012/2013), and the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiatives. This work was also supported by the RNA Biology Centre at the Cancer Science Institute of Singapore, NUS, as part of funding under the Singapore Ministry of Education’s Tier 3 grants (grant MOE2014-T3-1-006).
Authorship
Contribution: P.S. conceived the study, designed and performed experiments, analyzed the data, and wrote the manuscript; M.S., L.H., and D.K. performed experiments; A.M. and P.D. performed bioinformatics and statistical analyses and wrote the manuscript; W.W.T. maintained mouse colonies; M.C.L., M.F., O.A., and H.Y. performed 4C-seq experiments and analysis; J.S., and M.Z.H. generated the Δ+6 kb enh mice; V.M. and H.P.K. conceived and supervised the study, discussed and interpreted the data, and wrote the manuscript; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Pavithra Shyamsunder, Cancer Science Institute of Singapore, National University of Singapore, 14 Medical Dr, Singapore 117599; e-mail: csips@nus.edu.sg; and Vikas Madan, Cancer Science Institute of Singapore, National University of Singapore, 14 Medical Dr, Singapore 117599; e-mail: csivm@nus.edu.sg.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal