

Key Points
Combined 5-azacytidine and romidepsin are reasonably well tolerated in patients with relapsed/refractory lymphoid malignancies.
This combination produced overall and complete response rates of 73% and 55%, respectively, in patients with T-cell lymphoma.

Abstract
The peripheral T-cell lymphomas (PTCLs) are uniquely sensitive to epigenetic modifiers. Based on the synergism between histone deacetylase inhibitors and hypomethylating agents that we established in preclinical PTCL models, we conducted a phase 1 study of oral 5-azacytidine (AZA) and romidepsin (ROMI) in patients with advanced lymphoid malignancies, with emphasis on PTCL. According to a 3 + 3 design, patients were assigned to 1 of 7 cohorts with AZA doses ranging from 100 mg daily on days 1 to 14 to 300 mg daily on days 1 to 21, ROMI doses ranging from 10 mg/m2 on days 8 and 15 to 14 mg/m2 on days 8, 15, and 22, with cycles of 21 to 35 days. Coprimary end points included maximum tolerated dose (MTD) and dose-limiting toxicity (DLT). We treated a total of 31 patients. The MTD was AZA 300 mg on days 1 to 14 and ROMI 14 mg/m2 on days 8, 15, and 22 on a 35-day cycle. DLTs included grade 4 thrombocytopenia, prolonged grade 3 thrombocytopenia, grade 4 neutropenia, and pleural effusion. There were no treatment-related deaths. The combination was substantially more active in patients with PTCL than in those with non–T-cell lymphoma. The overall response rate in all, non–T-cell, and T-cell lymphoma patients was 32%, 10%, and 73%, respectively, and the complete response rates were 23%, 5%, and 55%, respectively. We did not find an association between response and level of demethylation or tumor mutational profile. This study establishes that combined epigenetic modifiers are potently active in PTCL patients. This trial was registered at www.clinicaltrials.gov as NCT01998035.
Introduction
Many converging lines of evidence suggest that peripheral T-cell lymphomas (PTCLs) may be prototypical epigenetic diseases. First, 4 histone deacetylase inhibitors (HDACis), vorinostat, romidepsin [ROMI], belinostat, and chidamide, have received regulatory approvals for patients with relapsed or refractory (R/R) PTCL. These drugs exhibit clear class effects, producing overall response rates (ORRs) of 25% to 35%, with highly favorable durations of response (DORs) across a diversity of PTCL subtypes. Second, mutations in genes governing epigenetic functions, including isocitrate dehydrogenase-2 (IDH2), ten-eleven translocation-2 (TET2), and DNA methyl transferase-3A (DNMT3A), are commonly found in PTCLs, particularly angioimmunoblastic T-cell lymphoma (AITL) and PTCL with a T-follicular helper phenotype. These mutations conspire to produce transcriptional silencing through DNA methylation. For example, mutant IDH2 converts isocitrate to R-2-hydroxyglutarate, which abrogates its function as a cofactor for oxidative demethylases like TET2 or lysine-specific demethylase family genes. Likewise, inactivating mutations in TET2 and DNMT3A are seen in patients with AITL and PTCL-not otherwise specified (NOS).1,2 In theory, hypomethylating agents would be the pharmacologic counterbalance to the biological consequences of these genetic and physiologic effects.
A third line of evidence comes from genetically manipulated murine models predicated on TET2 mutations, which induce development of spontaneous AITL.3,4 About half of all AITL cases carry a loss-of-function mutation in TET2, a tumor-suppressor gene. In addition, ∼70% of patients with AITL and ∼20% of those with PTCL-NOS carry a highly recurrent mutation (p.Gly17Val) in Ras homolog family member A (RHOA), a gene coding for a small GTPase.5-7 Recent studies in animal models revealed that RHOA G17V mutations can induce lymphomas with a T-follicular helper phenotype, alone8 and in cooperation with TET2 mutations.9 These results formally establish a unique cooperativity between epigenetic drivers and other recurring mutations in PTCL, which can lead to T-cell lymphomagenesis.
Finally, in 2 studies, our group was the first to show that combinations of HDACi and a host of other drugs active in PTCL, including hypomethylating agents, exhibit marked synergy in preclinical models of PTCL. Marchi et al,10 and more recently Scotto et al,11 established major class synergy between the HDACi and hypomethylating agents across a diverse panel of PTCL cell lines and in an in vivo xenograft model of T-cell lymphoma. Taken together, these data support the merits of an epigenetic platform comprising an HDACi and hypomethylating agent as a rational and novel approach to treat PTCL. We report herein the results from the first clinical trial to study the effects of oral 5-azacytidine (AZA) and ROMI in patients with R/R non-Hodgkin lymphoma and Hodgkin lymphoma, with a focus on patients with PTCL.
Methods
Study and patients
To be eligible for this multicenter phase 1 trial, patients were required to be ≥18 years of age, have an Eastern Cooperative Oncology Group performance status of ≤2, and have histologically confirmed R/R non-Hodgkin lymphoma or Hodgkin lymphoma, with no curative options. There was no limit to the number of prior therapies, and autologous (ASCT) and allogeneic (alloSCT) stem cell transplantations were allowed. Additional inclusion criteria included liver function tests (ie, aspartate aminotransferase [AST] or alanine aminotransferase [ALT]) ≤2 times the institutional upper limit of normal, total bilirubin ≤1.5 times the upper limit of normal, a creatinine clearance ≥50 mL/min, an absolute neutrophil count ≥1000 cells/μL, and a platelet count ≥75 000/μL. Exclusion criteria included exposure to chemotherapy or radiotherapy within 2 weeks of study entry or lack of recovery from adverse events (AEs), caused by to therapy administered more than 2 weeks earlier. Other exclusionary criteria included: concomitant investigational therapy, systemic steroids not stabilized (at least 5 days in advance) to an equivalent of ≤10 mg/day of prednisone at the time of study entry; active malignancy; HIV; hepatitis A, B, or C infection; pregnancy or nursing; or uncontrolled intercurrent illness.
The study was approved by all institutional review boards and was conducted according to the provisions of the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Practice. All patients enrolled gave informed consent.
Procedures
Enrollment followed a standard 3 + 3 dose-escalation design with assignment to 1 of 7 cohorts, with no intracohort dose escalation. The cohorts were expanded by 3 patients until the maximum tolerated dose (MTD) was reached. Treatment was given until disease progression, unacceptable toxicity, or withdrawal of consent. Prophylactic pegfilgrastim (6 mg subcutaneously, once) or filgrastim (300-480 μg subcutaneously, daily) were administered after cycle 1, as deemed necessary by the investigator. We allowed additional standard supportive treatments as clinically indicated, including antiemetics, antipyretics, antihistamines, analgesics, antibiotics, antivirals, and blood products.
AEs were evaluated using the Common Toxicity Criteria for Adverse Events, version 4.0, and monitored at each study visit. Laboratory tests were performed on day 1 of each cycle and included complete blood counts with differential and comprehensive metabolic profile. Dose-limiting toxicity (DLT) was assessed during cycle 1 only and was defined as any missed dose or toxicity at least possibly related to the study drug that occurred up to 7 days after completion of cycle 1 and resulted in a delay of initiation of cycle 2; any grade 5 AE; grade 4 neutropenia that did not resolve to grade 2 or lower within 7 days; grade 4 thrombocytopenia; febrile neutropenia (ie, absolute neutrophil count <1000 cells/μL with a single temperature of >38.3°C or a sustained temperature of ≥38°C for >1 hour); and any grade 3 or higher nonhematologic toxicity, excluding nausea, vomiting, diarrhea, or dehydration lasting longer than 48 hours in the setting of inadequate compliance with supportive care measures; grade 3 electrolyte abnormalities that responded to medical intervention; grade 3 acidosis or alkalosis that returned to grade 2 or lower within 48 hours with medical intervention; isolated asymptomatic ALT, AST, or amylase elevation lasting ≤5 days; or grade 3 hypercholesterolemia, hypertriglyceridemia, constipation, or fatigue.
Statistical analysis
The study was open label and employed a standard 3 + 3 dose-escalation design. The coprimary objectives were to identify the DLT of oral AZA and ROMI in cycle 1, as well as the MTD defined as the highest dose level at which less than one third of the cohort had a DLT in the first cycle of therapy. Secondary objectives included the ORR, defined as the sum of complete response (CR) and partial response; progression-free survival (PFS), defined as the time from enrollment until disease progression or death from any cause; and DOR. Investigator-assessed responses were based on the International Harmonization Project Group 2007 Revised Response Criteria.12 Computed tomography or positron emission tomography-computed tomography scans were performed after cycles 2 and 6. Scans were repeated every 3 to 6 months until relapse or start of the next therapy. Patients receiving at least 1 dose of the study drug were evaluable for toxicity, and all patients completing at least 1 cycle of therapy were evaluable for response and time-dependent analyses. Patients who went on to undergo an SCT were censored at that time.
PFS and DOR were estimated using the Kaplan-Meier method, group comparisons were made using a 2-sided log-rank test, and the estimated hazard ratio and 95% confidence interval (CI) were calculated using Cox regression. Overall survival was assessed post hoc. We assessed changes in biomarkers over time and estimated the odds ratio by using a linear model, given the small sample size. All analyses were performed with SAS software, v 9.4.
Exploratory analyses
Liquid chromatography-tandem mass spectrometry
Details regarding quantitation are provided elsewhere.13 In brief, ROMI was extracted from plasma with 500 µL acetonitrile/methanol (4:1). A liquid chromatography-tandem mass spectrometry analysis was performed using the Agilent 6410 Triple Quad MS connected to the Agilent 1290 Infinity ultra–high-performance liquid chromatograph (Agilent Technologies, Santa Clara, CA). Data acquisition and peak integration were performed with MassHunter software, v 3.1. The assay performance was validated according to US Food and Drug Administration guidelines.14 The lower limit of quantitation for ROMI was 2.5 ng/mL. The intra-assay precision and accuracy for ROMI were 3.9% and 101.9%, respectively, and the interassay precision was 3.0%. Technical issues prevented reliable AZA measurement in the phase 1 study.
Pharmacokinetic analysis
Plasma samples were collected on day 8 of cycle 1 at the following time points: before infusion and at 4, 4.5, 5, 6, 7, 28, and 52 hours. Noncompartmental analysis was performed with Phoenix Winnonlin software (Certara, Princeton, NJ) and included the usual parameters.
DNA methylation studies
Methylation assays are described in detail elsewhere.15 In brief, peripheral blood samples were collected before treatment, on days 8, 15, and 22 of cycle 1, and on day 1 of cycles 2 and 3. Methylation profiling was performed with the Infinium Methylation EPIC BeadChip, as described by the manufacturer (Illumina; San Diego, CA). Global DNA methylation scores (GDMSs) were derived for each sample by calculating the percentage of methylated loci with β > 0.7. Differential methylation for each CpG site was performed on M-values using the limma R package. Values below 5% false discovery rate were deemed significant. Rows were clustered using the Pearson correlation and Ward linkage. The R package chromoMap was used to represent the chromosomal location of differentially methylated regions.
Next-generation sequencing
Details regarding next-generation sequencing (NGS) have been presented elsewhere.16 In brief, NGS was performed with a custom panel of 465 cancer-associated genes. Genomic DNA was extracted using the QIAmp DNA Mini Kit or the QIAmp DNA FFPE kit (Qiagen, Germantown, MD) and fragmented. Sequencing was performed with paired-end sequencing chemistry (Illumina HiSeq2500 using Illumina TruSeq, v3; San Diego, CA). Mapping and alignment were performed with NextGene Software (Softgenetics, State College, PA). Germline polymorphisms were excluded after cross-referencing to the population databases (ExAC Browser, 1000 Genomes Project, and Exome Variant Server).
Results
Patients and treatment
In toto, 33 patients were screened for eligibility. From November 2013 through January 2016, 28 patients were screened for eligibility in the phase 1 study, with 26 being enrolled (1 screening failure was due to elevated transaminases and 1 to lack of evaluable disease). From April 2017 through October 2017, 5 additional patients with PTCL were enrolled in an expansion cohort treated at the MTD. Pretreatment characteristics of the per-protocol population are described in Table 1. Overall, the population was heavily pretreated, with a median of 6 prior lines of therapy (range, 1-15). All patients had received standard and accepted therapy. Fourteen (54%) and 5 (19%) patients had undergone ASCT and alloSCT, respectively.
Demographic characteristics of patients enrolled in the phase 1 study plus the expansion cohort of patients with peripheral T-cell lymphoma
| Characteristic | Data (N = 31) |
|---|---|
| Age, median (range), y | 57 (23-79) |
| Sex, n (%) | |
| Male | 19 (61) |
| Female | 12 (39) |
| Ethnicity, n (%) | |
| White, non-Hispanic | 19 (61) |
| White, Hispanic | 6 (20) |
| Black, non-Hispanic | 1 (3) |
| Black, Hispanic | 4 (13) |
| Asian or Pacific Islander | 1 (3) |
| Lymphoma subtype, n (%) | |
| Hodgkin lymphoma | 12 (39) |
| Diffuse large B-cell lymphoma* | 6 (20) |
| Follicular lymphoma | 2 (6) |
| T-cell lymphoma | 11 (35) |
| Angioimmunoblastic T-cell lymphoma | 3 (10) |
| Adult T-cell leukemia/lymphoma | 2 (6) |
| Peripheral T-cell lymphoma-NOS (cutaneous) | 1 (3) |
| Anaplastic large cell lymphoma, cutaneous | 1 (3) |
| Cutaneous T-cell lymphoma | 1 (3) |
| Enteropathy-associated T-cell lymphoma | 1 (3) |
| Extranodal natural killer/T-cell lymphoma | 1 (3) |
| T-cell lymphoblastic lymphoma | 1 (3) |
| Median number of prior therapies | 6 (1-15) |
| Prior regimens (at least 1 cycle), n (%) | |
| Hodgkin lymphoma (n = 12) | |
| ABVD-like, BEACOPP | 12 (39) |
| Platinum-based (ICE, ESHAP) | 12 (39) |
| Bv-based (Bv, Bv+B, Bv+AVD) | 12 (39) |
| Other alkylator-based (COPP, MOPP, B) | 11 (35) |
| Lenalidomide | 8 (26) |
| Gem-based (gem or GND) | 7 (23) |
| Experimental drug† | 7 (23) |
| Other therapy‡ | 6 (20) |
| B-cell lymphoma (n = 8) | |
| Anthracycline-based (R-CHOP, R-EPOCH) | 7 (23) |
| Platinum-based (R-ICE, R-DICE) | 6 (20) |
| Nonanthracycline-based (R-CVP, R-FND, R-B, R-B-bortezomib, R-F) | 4 (13) |
| Single agent anti-CD20 antibody (rituximab, ublituximab) | 4 (13) |
| Idelalisib + rituximab | 2 (6) |
| Experimental drug or other therapy§ | 5 (16) |
| T-cell lymphoma (n = 11) | |
| Anthracycline-based (CHOP, CHOEP, EPOCH±bortezomib) | 8 (26) |
| Gem-based (gem, p-gemox, gem+liposomal cytarabine) | 4 (13) |
| HDAC inhibitor (vorinostat, ROMI) | 3 (10) |
| Bv | 2 (6) |
| Pralatrexate | 1 (3) |
| Experimental drug or other therapy, n (%)‖ | 6 (20) |
| Prior ASCT, n (%) | 16 (52) |
| Prior alloSCT, n (%) | 5 (16) |
| Radiotherapy/radioimmunotherapy, n (%) | 10 (32) |
| Characteristic | Data (N = 31) |
|---|---|
| Age, median (range), y | 57 (23-79) |
| Sex, n (%) | |
| Male | 19 (61) |
| Female | 12 (39) |
| Ethnicity, n (%) | |
| White, non-Hispanic | 19 (61) |
| White, Hispanic | 6 (20) |
| Black, non-Hispanic | 1 (3) |
| Black, Hispanic | 4 (13) |
| Asian or Pacific Islander | 1 (3) |
| Lymphoma subtype, n (%) | |
| Hodgkin lymphoma | 12 (39) |
| Diffuse large B-cell lymphoma* | 6 (20) |
| Follicular lymphoma | 2 (6) |
| T-cell lymphoma | 11 (35) |
| Angioimmunoblastic T-cell lymphoma | 3 (10) |
| Adult T-cell leukemia/lymphoma | 2 (6) |
| Peripheral T-cell lymphoma-NOS (cutaneous) | 1 (3) |
| Anaplastic large cell lymphoma, cutaneous | 1 (3) |
| Cutaneous T-cell lymphoma | 1 (3) |
| Enteropathy-associated T-cell lymphoma | 1 (3) |
| Extranodal natural killer/T-cell lymphoma | 1 (3) |
| T-cell lymphoblastic lymphoma | 1 (3) |
| Median number of prior therapies | 6 (1-15) |
| Prior regimens (at least 1 cycle), n (%) | |
| Hodgkin lymphoma (n = 12) | |
| ABVD-like, BEACOPP | 12 (39) |
| Platinum-based (ICE, ESHAP) | 12 (39) |
| Bv-based (Bv, Bv+B, Bv+AVD) | 12 (39) |
| Other alkylator-based (COPP, MOPP, B) | 11 (35) |
| Lenalidomide | 8 (26) |
| Gem-based (gem or GND) | 7 (23) |
| Experimental drug† | 7 (23) |
| Other therapy‡ | 6 (20) |
| B-cell lymphoma (n = 8) | |
| Anthracycline-based (R-CHOP, R-EPOCH) | 7 (23) |
| Platinum-based (R-ICE, R-DICE) | 6 (20) |
| Nonanthracycline-based (R-CVP, R-FND, R-B, R-B-bortezomib, R-F) | 4 (13) |
| Single agent anti-CD20 antibody (rituximab, ublituximab) | 4 (13) |
| Idelalisib + rituximab | 2 (6) |
| Experimental drug or other therapy§ | 5 (16) |
| T-cell lymphoma (n = 11) | |
| Anthracycline-based (CHOP, CHOEP, EPOCH±bortezomib) | 8 (26) |
| Gem-based (gem, p-gemox, gem+liposomal cytarabine) | 4 (13) |
| HDAC inhibitor (vorinostat, ROMI) | 3 (10) |
| Bv | 2 (6) |
| Pralatrexate | 1 (3) |
| Experimental drug or other therapy, n (%)‖ | 6 (20) |
| Prior ASCT, n (%) | 16 (52) |
| Prior alloSCT, n (%) | 5 (16) |
| Radiotherapy/radioimmunotherapy, n (%) | 10 (32) |
A(B)VD, Adriamycin, bleomycin, vinblastine, dacarbazine; B bendamustine; BAC, bendamustine, cytarabine; BEACOPP, bleomycin, etoposide, Adriamycin, cyclophosphamide, vincristine, procarbazine, prednisone; Bv, brentuximab vedotin; COPP, cyclophosphamide, vincristine, procarbazine, prednisone/Adriamycin; CVP, cyclophosphamide, vincristin, prednisone; (D)ICE, (dexamethasone) ifosphamide, carboplatin, etoposide; EPOCH, etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin; ESHAP, etoposide, methylprednisolone, high-dose cytarabine, cisplatin; FND, fludarabine, mitoxanthrone, dexamethasone; Gem, gemcitabine; gemox, gemcitabine+oxaliplatin; GND, gemcitabine, navelbine, liposomal doxorubicin; IVAC, etoposide, ifosfamide, cytarabine, methotrexate; MOPP, mechlorethamine, vincristine, procarbazine, prednisone; (R)-CHO(E)P, rituximab, cyclophosphamide, doxorubicin, vincristine (etoposide) prednisone; (p)GemOx, (peg-asparaginase) gemcitabine, oxaliplatin; SMILE, dexamethasone, methotrexate, ifosfamide, l-asparaginase, etoposide.
3 nodal, 2 transformed from follicular lymphoma, 1 cutaneous.
ACY1215, umbralisib, PI3K inhibitor+JAK inhibitor, pralatrexate+ROMI, vorinostat+niacinamide, Nedd8-activating enzyme inhibitor MLN4924, CUDC907, belinostat, CPI-0610, SB743921, MDX-060.
EPOCH, IVAC, vinblastine+bleomycin+methotrexate, vinblastine, pembrolizumab (compassionate use), EBV-specific cytotoxic T-lymphocytes, alternative medicine.
Anti-CD37, vorinostat-niacinamide±etoposide, ibrutinib, umbralisib, pralatrexate, brentuximab vedotin, R-lenalidomide.
Pralatrexate+ROMI, bortezomib, Nelarabine, mogamulizumab, bexarotene, photopheresis±interferon, total skin electron beam therapy, pembrolizumab, bendamustine, ICE, SMILE, BAC.
Patient disposition is illustrated in the Consolidated Standards of Reporting Trials diagram (Figure 1). Three patients were enrolled in cohort 1 without a DLT, followed by 3 patients in cohort 2, of whom 2 experienced DLTs (ie, thrombocytopenia and pleural effusion). After cohort 2, we extended the cycle duration to 28 days. Three patients were then enrolled in cohort 3 with no DLT. Subsequent accrual occurred as follows: 3 patients in cohort 4, 3 in cohort 5, and 3 in cohort 6, with no DLTs. Cohort 7 produced DLTs in 1 of the first 3 patients (grade 4 neutropenia), prompting cohort expansion, and in 2 of the second 3 (thrombocytopenia leading to missed doses of study drug and sepsis). Thus, the cohort 7 dose was declared the maximum administrable, and the cohort 6 dose (ie, oral AZA 300 mg orally daily on days 1 to 14 and ROMI 14 mg/m2 intravenously on days 8, 15, and 22, on a 35-day cycle) was declared the MTD and recommended phase 2 dose (RP2D; supplemental Material, available on the Blood Web site).

Diagram of study patient disposition. *Identified as the maximum tolerated dose, used in the dose expansion cohort. †Identified as the maximum administered dose.
Diagram of study patient disposition. *Identified as the maximum tolerated dose, used in the dose expansion cohort. †Identified as the maximum administered dose.
Safety
A total of 133 cycles (0.5-16 cycles per patient) have been administered at the time of this writing. No patient had dose reductions and, 2 patients permanently discontinued therapy due to toxicity (1 with nausea and vomiting and 1 with sepsis). No grade 5 toxicities occurred. Treatment-emergent AEs are described in Table 2. The most common grade ≥3 AEs were cytopenias, particularly thrombocytopenia and lymphopenia. Other notable AEs of grade 3 or higher included hypotension (12% of patients), febrile neutropenia (8%), hyponatremia (8%), and sepsis (4%). All patients experienced grade 1 and 2 AEs, but these were mostly transient and readily manageable (Table 2). Nausea and vomiting were relatively common, albeit almost exclusively of grade 1 or 2 severity and occurring mostly during the first few days of each cycle. These AEs were largely attributed to the AZA tablets. The effective antinausea regimen was as follows: during treatment with oral AZA, patients received granisetron 1 mg orally every 12 hours (later replaced with transdermal granisetron 3.1 mg every 24 hours) and metoclopramide 5 mg orally 30 minutes before dosing; in addition, patients received dexamethasone 12 mg intravenously and ondansetron 16 mg intravenously 30 minutes before each ROMI dose. In virtually all cases, nausea and vomiting disappeared during cycles 2 and 3, at which point patients were treated with oral ondansetron on an as-needed basis. Among 8 patients treated at the RP2D, major grades 3 and 4 treatment-emergent AEs included thrombocytopenia, neutropenia, and infections, including 1 case of febrile neutropenia (Table 2).
Treatment-emergent AEs recorded in the phase 1 population and in patients treated at the RP2D
| AE | Phase 1 population (n = 26), n (%) | Patients treated at the RP2D* (n = 8), n (%) | ||
|---|---|---|---|---|
| Grades 1-2 | Grades 3-4 | Grades 1-2 | Grades 3-4 | |
| Alkaline phosphatase elevation | 9 (35) | 1 (4) | 2 (25) | 0 |
| Anemia | 12 (46) | 5 (19) | 5 (62) | 1 (12) |
| Anorexia | — | — | 2 (25) | 0 |
| AST elevation | — | — | 2 (25) | 0 |
| Constipation | — | — | 3 (37) | 0 |
| Cough | 3 (12) | — | 4 (50) | 0 |
| Creatinine elevation | 8 (31) | — | 3 (37) | 0 |
| Diarrhea | 7 (27) | — | 2 (25) | 1 (12) |
| Fatigue | 10 (38) | 1 (4) | 3 (37) | 0 |
| Febrile neutropenia | — | 2 (8) | — | 1 (12) |
| Fever | 3 (12) | — | 3 (37) | 1 (12) |
| General disorders and administration site conditions | 4 (15) | — | 2 (25) | 0 |
| Hiccups | — | — | 2 (25) | 0 |
| Hyperbilirubinemia | — | — | — | 1 (12) |
| Hypercalcemia | 3 (12) | — | — | — |
| Hyperchloremia | — | — | 2 (25) | 0 |
| Hyperglycemia | 21 (81) | 1 (4) | 7 (87) | 1 (12) |
| Hypermagnesemia | 1 (4) | 1 (4) | — | — |
| Hypoalbuminemia | 13 (50) | — | 4 (50) | 0 |
| Hypocalcemia | 5 (19) | — | — | — |
| Hypochloremia | 5 (19) | — | 3 (37) | 0 |
| Hypogammaglobulinemia | 8 (31) | — | — | — |
| Hypoglycemia | 3 (12) | — | — | — |
| Hypokalemia | 4 (15) | 1 (4) | 2 (25) | — |
| Hyponatremia | 12 (46) | 2 (8) | 2 (25) | 0 |
| Hypophosphatemia | — | — | 2 (25) | 0 |
| Hypoproteinemia | 17 (65) | — | 5 (62) | 0 |
| Hypotension | — | 3 (12) | — | — |
| Insomnia | — | — | 2 (25) | 0 |
| Lung infection | — | — | — | 2 (25) |
| Lymphocyte count decrease | 10 (38) | 11 (42) | 3 (37) | 2 (25) |
| Mucositis oral | 4 (15) | — | 2 (25) | 0 |
| Nausea | 14 (54) | 1 (4) | 4 (50) | 0 |
| Neutrophil count decrease | 6 (23) | 11 (42) | 1 (12) | 3 (37) |
| Pain | 3 (12) | — | — | — |
| Platelet count decrease | 14 (54) | 7 (27) | 2 (25) | 5 (62) |
| Pleural effusion | — | 1 (4) | — | — |
| Urinary tract infection | — | 1 (4) | — | — |
| Vomiting | 12 (46) | — | 3 (37) | 0 |
| White blood cell count decrease | 13 (50) | 6 (23) | 4 (50) | 2 (25) |
| AE | Phase 1 population (n = 26), n (%) | Patients treated at the RP2D* (n = 8), n (%) | ||
|---|---|---|---|---|
| Grades 1-2 | Grades 3-4 | Grades 1-2 | Grades 3-4 | |
| Alkaline phosphatase elevation | 9 (35) | 1 (4) | 2 (25) | 0 |
| Anemia | 12 (46) | 5 (19) | 5 (62) | 1 (12) |
| Anorexia | — | — | 2 (25) | 0 |
| AST elevation | — | — | 2 (25) | 0 |
| Constipation | — | — | 3 (37) | 0 |
| Cough | 3 (12) | — | 4 (50) | 0 |
| Creatinine elevation | 8 (31) | — | 3 (37) | 0 |
| Diarrhea | 7 (27) | — | 2 (25) | 1 (12) |
| Fatigue | 10 (38) | 1 (4) | 3 (37) | 0 |
| Febrile neutropenia | — | 2 (8) | — | 1 (12) |
| Fever | 3 (12) | — | 3 (37) | 1 (12) |
| General disorders and administration site conditions | 4 (15) | — | 2 (25) | 0 |
| Hiccups | — | — | 2 (25) | 0 |
| Hyperbilirubinemia | — | — | — | 1 (12) |
| Hypercalcemia | 3 (12) | — | — | — |
| Hyperchloremia | — | — | 2 (25) | 0 |
| Hyperglycemia | 21 (81) | 1 (4) | 7 (87) | 1 (12) |
| Hypermagnesemia | 1 (4) | 1 (4) | — | — |
| Hypoalbuminemia | 13 (50) | — | 4 (50) | 0 |
| Hypocalcemia | 5 (19) | — | — | — |
| Hypochloremia | 5 (19) | — | 3 (37) | 0 |
| Hypogammaglobulinemia | 8 (31) | — | — | — |
| Hypoglycemia | 3 (12) | — | — | — |
| Hypokalemia | 4 (15) | 1 (4) | 2 (25) | — |
| Hyponatremia | 12 (46) | 2 (8) | 2 (25) | 0 |
| Hypophosphatemia | — | — | 2 (25) | 0 |
| Hypoproteinemia | 17 (65) | — | 5 (62) | 0 |
| Hypotension | — | 3 (12) | — | — |
| Insomnia | — | — | 2 (25) | 0 |
| Lung infection | — | — | — | 2 (25) |
| Lymphocyte count decrease | 10 (38) | 11 (42) | 3 (37) | 2 (25) |
| Mucositis oral | 4 (15) | — | 2 (25) | 0 |
| Nausea | 14 (54) | 1 (4) | 4 (50) | 0 |
| Neutrophil count decrease | 6 (23) | 11 (42) | 1 (12) | 3 (37) |
| Pain | 3 (12) | — | — | — |
| Platelet count decrease | 14 (54) | 7 (27) | 2 (25) | 5 (62) |
| Pleural effusion | — | 1 (4) | — | — |
| Urinary tract infection | — | 1 (4) | — | — |
| Vomiting | 12 (46) | — | 3 (37) | 0 |
| White blood cell count decrease | 13 (50) | 6 (23) | 4 (50) | 2 (25) |
For the phase 1 population, grade 1 and 2 AEs occurring in more than 10 of patients are reported, regardless of attribution to study drugs. For patients treated at the RP2D, grade 1 and 2 AEs occurring in at least 2 patients are reported, regardless of attribution to study drugs.
This group includes 3 patients enrolled in the phase 1 trial and the 5 patients treated in the exploratory cohort.
Efficacy
Overall, 10 (32%) patients achieved an objective response, and 7 (23%) attained a CR. The combination of oral AZA and ROMI was significantly more active in patients with PTCL than in those with non–T-cell lymphomas. Only 2 of 20 patients with non–T-cell lymphoma responded (10%), only one of whom achieved a CR (5%). Among 11 patients with PTCL (6 in the phase 1 study and 5 in the expansion cohort), 8 (73%) achieved a response, with 6 (55%) attaining a CR (Table 3), and those differences were statistically significant (ORR, P = .001; CR, P = .01). The individual best response of each patient is presented in the supplemental Material.
Response to oral 5-AZA and ROMI across study subpopulations
| Response type | All patients (N = 31), n (%) | Phase 1 population (n = 26), n (%) | Expansion cohort (n = 5), n (%) | Non–T-cell lymphoma (n = 20), n (%) | T-cell lymphoma (n = 11), n (%) |
|---|---|---|---|---|---|
| Overall response | 10 (32) | 6 (23) | 4 (80) | 2 (10) | 8 (73) |
| Complete response | 7 (23) | 3 (12) | 4 (80) | 1 (5) | 6 (55) |
| Partial response | 3 (10) | 3 (12) | 0 | 1 (5) | 2 (18) |
| Stable disease | 7 (23) | 7 (27) | 0 | 7 (35) | 0 |
| Progressive disease | 11 (35) | 10 (38) | 1 (20) | 9 (45) | 2 (18) |
| Not evaluable | 3 (10) | 3 (12) | 0 | 2 (10) | 1 (9) |
| Response type | All patients (N = 31), n (%) | Phase 1 population (n = 26), n (%) | Expansion cohort (n = 5), n (%) | Non–T-cell lymphoma (n = 20), n (%) | T-cell lymphoma (n = 11), n (%) |
|---|---|---|---|---|---|
| Overall response | 10 (32) | 6 (23) | 4 (80) | 2 (10) | 8 (73) |
| Complete response | 7 (23) | 3 (12) | 4 (80) | 1 (5) | 6 (55) |
| Partial response | 3 (10) | 3 (12) | 0 | 1 (5) | 2 (18) |
| Stable disease | 7 (23) | 7 (27) | 0 | 7 (35) | 0 |
| Progressive disease | 11 (35) | 10 (38) | 1 (20) | 9 (45) | 2 (18) |
| Not evaluable | 3 (10) | 3 (12) | 0 | 2 (10) | 1 (9) |
Tumor burden was measurable in 25 patients. Sixteen patients (64%) experienced some degree of tumor shrinkage, with all 3 AITL patients achieving 100% tumor volume reduction (Figure 2A, inset). Among the 7 complete responders, 4 achieved a CR at the first assessment (after 2 cycles), whereas 3 achieved a partial response, which converted to CR at the subsequent evaluation (after 6 cycles; Figure 2B). At a follow-up of 15.3 months (range, 1.6-54.3) the median PFS for all patients was 3.6 months (95% CI, 1.9-5.3). The median PFS was not reached for patients with PTCL and lasted longer, compared with those with non–T-cell lymphomas (2.5 months; 95% CI, 0.4-4.6; Figure 3A). The median DOR was not reached (range, 1.8-16.3+ months), and 6 patients with T-cell lymphoma who achieved a response went on to undergo SCT and were censored at that time (Figure 3B).
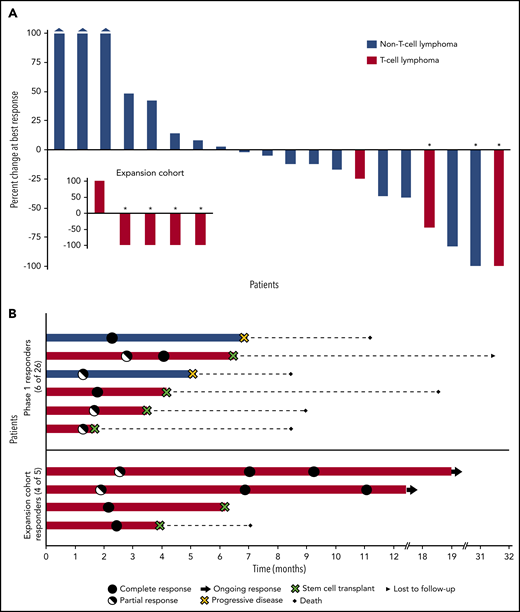
Waterfall and swimmers plots. (A) Percentage change in tumor burden. The waterfall plot depicts the percentage change in tumor volume at the time of best response for 20 evaluable patients in the phase 1 population and the 5 exploratory patients with T-cell lymphoma (inset). Three patients had evaluable, but not measurable disease: 1 patient had CD8+ cutaneous peripheral T-cell lymphoma, 1 had cutaneous anaplastic large cell lymphoma, and 1 had cutaneous diffuse large B-cell lymphoma; all achieved a partial response. Each bar represents an individual patient. Asterisks indicate CR. The 3 leftmost bars of the main plot represent patients whose tumor size increased by 100% or more. (B) Duration of response. For patients who underwent stem cell transplantation, the duration of response was censored at the time of this procedure. the figure highlights how the responder population is enriched in patients with T-cell lymphoma who may also exhibit longer lasting responses to the combination of 5-AZA and ROMI compared with their non–T-cell lymphoma counterparts.
Waterfall and swimmers plots. (A) Percentage change in tumor burden. The waterfall plot depicts the percentage change in tumor volume at the time of best response for 20 evaluable patients in the phase 1 population and the 5 exploratory patients with T-cell lymphoma (inset). Three patients had evaluable, but not measurable disease: 1 patient had CD8+ cutaneous peripheral T-cell lymphoma, 1 had cutaneous anaplastic large cell lymphoma, and 1 had cutaneous diffuse large B-cell lymphoma; all achieved a partial response. Each bar represents an individual patient. Asterisks indicate CR. The 3 leftmost bars of the main plot represent patients whose tumor size increased by 100% or more. (B) Duration of response. For patients who underwent stem cell transplantation, the duration of response was censored at the time of this procedure. the figure highlights how the responder population is enriched in patients with T-cell lymphoma who may also exhibit longer lasting responses to the combination of 5-AZA and ROMI compared with their non–T-cell lymphoma counterparts.

Progression free and overall survival. PFS (A) and DOR (B) in patients with T-cell lymphoma and those with non–T-cell-lymphoma.
Progression free and overall survival. PFS (A) and DOR (B) in patients with T-cell lymphoma and those with non–T-cell-lymphoma.
Correlative studies
We performed first-dose pharmacokinetic (PK) analysis on 25 patients in the phase 1 trial and on the 5 patients in the expansion cohort. Full PK parameters and the average plasma ROMI concentration over time are provided in the supplemental Material. The maximum plasma concentration (Cmax) of ROMI (14 mg/m2), coadministered with oral AZA (300 mg), was 460.20 ng/mL (0.85 µM). Notably, the in vitro 50% inhibitory concentration (IC50) of ROMI in T-cell lymphoma cell lines ranged from 1.2 to 1.6 nM; thus, the plasma concentration of ROMI administered at the RP2D well exceeded the IC50. These data are comparable to those published in other experiences of single-agent ROMI at 14 mg/m2,17-19 suggesting no significant PK effect of oral AZA on ROMI.
We performed longitudinal, genome-wide DNA methylation profiling in the 5 PTCL patients treated in the expansion cohort. Overall, the GDMS decreased in 4 of those 5 patients by cycle 1, day 8, compared with baseline (P = .026, across all DNA methylation sites). In this cohort, the GDMS did not appear to correlate with the clinical response (Figure 4A). We also analyzed methylation patterns at the single-nucleotide level, including CpG and non-CpG loci in both genic and nongenic regions. Interestingly, demethylation was observed at all tested genomic regions in 4 patients, regardless of the baseline methylation level (supplemental Material). In an attempt to identify a common response signature for oral AZA and ROMI across patients, we then examined the differentially methylated patterns at CpG loci between baseline and each posttreatment time point (Figure 4B-C). Our analyses revealed 5 significantly differentially demethylated chromosomal regions at cycle 1, day 15, and 24 regions at cycle 2, day 22 (Figure 4B; supplemental Material). A hierarchical clustering analysis of these sites revealed that the methylation status of several regions changed after treatment in each patient (Figure 4C), suggesting that the pharmacodynamic effects of oral AZA were attained in all patients.
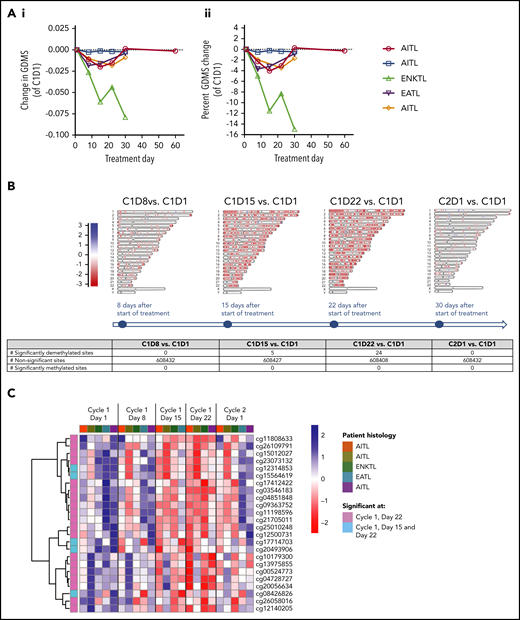
DNA methylation change in peripheral blood of the 5 PTCL patients treated in the expansion cohort. (Ai) Absolute and (Aii) percentage change in GDMS values compared with cycle 1, day 1. All 3 patients with AITL, including 1 with no appreciable demethylation, responded, 1 patient with extranodal natural killer/T cell lymphoma with profound demethylation responded, and 1 patient with enteropathy-associated intestinal T-cell lymphoma with nearly 40% reduction in GDMS had progression of disease. (B) Time course of global blood DNA methylation changes compared with day 1 of cycle 1 across all chromosomes after treatment; only sites with an absolute log-fold change >1 are shown. Methylation values are color coded such that red indicates lower methylation and blue higher methylation. (C) Heatmap of significantly differentially methylated CpG sites from day 1 of cycle 1 to day 1 of cycle 2. The leftmost column indicates significantly demethylated CpG site on day 22 (pink), and on days 15 and 22 (light blue) at values below a false discovery rate of 5%. The top row indicates patient histology. Methylation values scaled by CpG site are color coded such that red indicates lower methylation and blue higher methylation. ENKTL, extranodal NK/T-cell lymphoma; EATL, enteropathy-associated T-cell lymphoma.
DNA methylation change in peripheral blood of the 5 PTCL patients treated in the expansion cohort. (Ai) Absolute and (Aii) percentage change in GDMS values compared with cycle 1, day 1. All 3 patients with AITL, including 1 with no appreciable demethylation, responded, 1 patient with extranodal natural killer/T cell lymphoma with profound demethylation responded, and 1 patient with enteropathy-associated intestinal T-cell lymphoma with nearly 40% reduction in GDMS had progression of disease. (B) Time course of global blood DNA methylation changes compared with day 1 of cycle 1 across all chromosomes after treatment; only sites with an absolute log-fold change >1 are shown. Methylation values are color coded such that red indicates lower methylation and blue higher methylation. (C) Heatmap of significantly differentially methylated CpG sites from day 1 of cycle 1 to day 1 of cycle 2. The leftmost column indicates significantly demethylated CpG site on day 22 (pink), and on days 15 and 22 (light blue) at values below a false discovery rate of 5%. The top row indicates patient histology. Methylation values scaled by CpG site are color coded such that red indicates lower methylation and blue higher methylation. ENKTL, extranodal NK/T-cell lymphoma; EATL, enteropathy-associated T-cell lymphoma.
NGS data were available in 9 PTCL patients. Three of them (2 with AITL and 1 with extranodal NK cell lymphoma) had a TET2 mutation, and 1 of these had a coexisting DNMT3A mutation. IDH2 mutations were not found. All patients with TET2 mutation and 3 of the remaining 6 had an objective response (supplemental Material).
Discussion
The consistent and reproducible activities of HDACi in PTCL, coupled with recurrent defects in methylation found in select subtypes, suggest that the PTCL may be more vulnerable to epigenetic modifier–based treatments compared with other malignant disorders. Indeed, the PTCL are uniquely vulnerable to HDACi, suggesting a potential lineage specificity of these agents. Despite consistent activity across different classes of HDACi, their mechanism of action remains elusive. In addition, the finding of recurring mutations in epigenetic regulators like TET2, IDH2, and DNMT3A raises the prospect that hypomethylating agents play a therapeutic role in PTCL, despite not having a major role in treating any other type of lymphoma. Although these defects in methylation are not pervasive throughout all subtypes of PTCL, they appear to be enriched in select subtypes such as AITL and PTCL-NOS.5 Presently, it is unclear to what extent these mutations portend responsiveness to hypomethylating agents, though it is likely that a broader spectrum of epigenetic dysregulation in PTCL influences this vulnerability beyond TET2, IDH2, or DNMT3. Certainly, the synergy between hypomethylating agents and HDACi observed in cell lines that are not known to carry these mutations suggests that the combination may target a broader spectrum of epigenetic biology independent of TET2, IDH2, and DNMT3 mutations.
The MTD in this phase 1 study was 300 mg oral AZA daily on days 1 to 14 and ROMI 14 mg/m2 on days 8, 15, and 22 on a 35-day cycle. Most trials seeking to exploit the hypomethylating effect of decitabine and AZA have focused on lower doses and concentrations, which lead to degradation of the DNA-DNMT complex, with hypomethylation of CpG islands without the concomitant DNA fragmentation seen at higher concentrations. PK studies of AZA confirmed that doses of 300 mg orally daily can achieve Cmax in the range of 401 to 507 nM,20 whereas the doses of ROMI produce concentrations well above the IC50 of the drug. In cell lines, concentrations of AZA between 100 and 800 nM appear to produce significant demethylation without DNA fragmentation.
The most common AEs included grade 3 or higher thrombocytopenia and neutropenia. The cytopenias were rapidly reversible and were not cumulative over time. Other AEs included nausea and vomiting, which were seen in most patients. Implementation of an effective antinausea regimen substantially reduced the severity and duration of these AEs.
The overall and CR rates of the combination across the entire study population were 32% and 23%, respectively. There was a significant lineage selectivity for the combination, where the ORR and CR in PTCL were 73% and 55%, respectively, compared with 10% and 5%, respectively, in non–T-cell lymphomas. PTCL responses proved durable, with 2 patients still in treatment and in CR after 18.9 and 12.5 months. Such high CR rates in a heavily pretreated population is noteworthy, as failure to achieve CR is a recognized impediment to undergoing ASCT. It is also notable that all patients with AITL achieved a CR, which may suggest exquisite vulnerability of this subtype to the oral AZA-ROMI combination. However, the small AITL sample size precludes firmer conclusions in this regard.
PK analyses suggested no direct influence of oral AZA on the metabolism of ROMI in terms of area under the curve and Cmax, based on previous experience with this single agent.17-19 Other PK studies analyzing the Cmax of oral, subcutaneous, and intravenous AZA were conducted after a single dose and showed significant intra- and interstudy variability,21,22 thus precluding an informed comparison between oral and parenteral formulations of the drug. However, although the DNA methylation patterns were heterogenous, the extent of GDMS changes with oral AZA was concordant with the one previously reported in patients with myelodysplastic syndrome or acute myeloid leukemia treated with the same schedule of oral AZA,20 and concurrent HDAC inhibition does not appear to interfere with this pharmacodynamic effect. We identified several significantly demethylated regions across the genome after oral AZA and ROMI. Whether genes in these chromosomal regions are involved in the mechanism of action of AZA-ROMI remains to be determined.
Delarue et al23 previously suggested that patients with PTCL who responded to injectable AZA may be enriched in TET2-mutated cases. In the small group of patients for whom NGS data were available, we did not observe a clear correlation between the presence of TET2 mutations and a clinical response to oral AZA and ROMI therapy. Because of the relatively small number of patients in this and other experiences, it is unclear whether the presence of TET2 mutations portends increased sensitivity to HDACi, hypomethylating agents, or a combination thereof.
In conclusion, we showed that the combination of oral AZA and ROMI is feasible and demonstrates excellent, nearly T-cell lineage-specific activity in patients with lymphoid malignancies. The high overall and CR rates observed make this regimen highly desirable for patients with R/R PTCL, particularly those for whom a SCT procedure is planned. Although the number of patients with T-cell lymphoma is small in this study, we have recently completed accrual to the phase 2 trial of this combination and will be validating these findings, including the methylation patterns and NGS, in a larger sample of patients with PTCL exclusively.
Deidentified individual participant data that underlie the results reported in this article will be made available, after deidentification. Data will be made available following publication and for up to 3 years thereafter. Interested individuals should submit a written request to the corresponding author. Only requests that contain a methodologically sound, clearly stated proposal will be considered. To gain access, interested requestors will need to sign a data access agreement.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Celgene Corporation, the Leukemia and Lymphoma Society, and the Lymphoma Research Fund of Columbia University for providing funding for this study.
Authorship
Contribution: O.A.O. conceived and designed the study; O.A.O., L.F., J.K.L., E.M., C.K., A.S., C.D., F.M., J.E.A., H.A.K., A.M.R., K.K., A.T.J., M.M., M.M.F., R.N., C.R.S., D.C.P., G.B., B.C., A.R., D.M., A.R.S., L. Sokol, and L. Scotto acquired the data; O.A.O. L.F., J.K.L., M.F., R.N., C.R.S., D.C.P., B.C., A.R., D.M., A.R.S., L. Sokol, and L. Scotto analyzed and interpreted the data; M.F.F. provided radiological interpretation; R.N., C.R.S., D.C.P., and G.B. provided correlative analyses; O.A.O., L.F., J.K.L., A.R., D.M., and C.R.S. wrote the paper; and B.C. performed the statistical analysis.
Conflict-of-interest disclosure: O.A.O. received grants from Celgene during the conduct of this study and personal fees (service on the data safety monitoring board) from Celgene, outside the submitted work. A.R.S. has received personal fees from Portola Pharmaceuticals and Kyowa-Hakko-Kirin and grants from Spectrum Pharmaceuticals outside the submitted work. L. Sokol has received personal fees from Celgene, Spectrum Pharmaceuticals, and Seattle Genetics outside the submitted work. J.E.A. received research grants from Appia Pharmaceuticals and honoraria from Janssen. A.S. has received honoraria from Seattle Genetics, Gilead Science, and Daiichi, and grants from Affimed, outside the submitted work. D.M. and A.R. are Celgene employees and own Celgene stock. The remaining authors declare no competing financial interests.
Correspondence: Owen A. O’Connor, Center for Lymphoid Malignancies, Department of Medicine, Columbia University Medical Center, 51 West 52st St, Suite 200, New York, NY 10019; e-mail: owenoconnor@cumc.columbia.edu.
