

Key Points
Isatuximab combined with pomalidomide/dexamethasone has a manageable safety profile with promising clinical activity.
Isatuximab 10 mg/kg (4 weekly doses followed by dosing every 2 weeks thereafter) has been selected for future combination studies.

Abstract
This phase 1b dose-escalation study evaluated isatuximab plus pomalidomide/dexamethasone in patients with relapsed/refractory multiple myeloma (RRMM). Patients who had received ≥2 prior MM therapies, including lenalidomide and a proteasome inhibitor (PI), were enrolled and received isatuximab at 5, 10, or 20 mg/kg (weekly for 4 weeks, followed by every 2 weeks), pomalidomide 4 mg (days 1-21), and dexamethasone 40 mg (weekly) in 28-day cycles until progression/intolerable toxicity. The primary objective was to determine the safety and recommended dose of isatuximab with this combination. Secondary objectives included evaluation of pharmacokinetics, immunogenicity, and efficacy. Forty-five patients received isatuximab (5 [n = 8], 10 [n = 31], or 20 [n = 6] mg/kg). Patients received a median of 3 (range, 1-10) prior lines; most were refractory to their last regimen (91%), with 82% lenalidomide-refractory and 84% PI-refractory. Median treatment duration was 9.6 months; 19 patients (42%) remain on treatment. Most common adverse events included fatigue (62%), and upper respiratory tract infection (42%), infusion reactions (42%), and dyspnea (40%). The most common grade ≥3 treatment-emergent adverse event was pneumonia, which occurred in 8 patients (17.8%). Hematologic laboratory abnormalities were common (lymphopenia, leukopenia, anemia, 98% each; neutropenia, 93%; and thrombocytopenia, 84%). Overall response rate was 62%; median duration of response was 18.7 months; median progression-free survival was 17.6 months. These results demonstrate potential meaningful clinical activity and a manageable safety profile of isatuximab plus pomalidomide/dexamethasone in heavily pretreated patients with RRMM. The 10 mg/kg weekly/every 2 weeks isatuximab dose was selected for future studies. This trial was registered at www.clinicaltrials.gov as #NCT02283775.
Introduction
Despite the availability of numerous therapies, multiple myeloma (MM) remains a burdensome and incurable disease for the vast majority of patients.1-3 The introduction of novel agents has improved response and survival rates in patients with MM; however, almost all patients will still relapse during or after completion of these treatments.4 For those patients who have received 2 or more prior lines of therapy, and are refractory to a proteasome inhibitor (PI) and an immunomodulatory drug (IMiD) (relapsed/refractory MM [RRMM]), pomalidomide is indicated but prognosis is poor and overall survival (OS) is short (median, 12.7 months).4,5 Therefore, new treatments are necessary to prolong OS and improve quality of life.
CD38 is highly expressed in MM, with nearly all malignant plasma cells from patients with MM showing surface expression of this protein,6,7 thus making CD38 an attractive target for anti-MM therapies. Our preclinical studies have demonstrated that isatuximab has a novel, direct mechanism of anti-MM activity, as well as targeting both constitutive and inducible T regulatory cells, thereby enhancing effector cell anti-MM activity.7-9 In addition, preclinical investigations have demonstrated that anti-CD38 therapies in combination treatment with IMiDs, including lenalidomide and pomalidomide, increase anti-MM activity.9,10 Furthermore, the anti-CD38 monoclonal antibody daratumumab has demonstrated efficacy as monotherapy in heavily pretreated patients with RRMM,11 which was enhanced when daratumumab was combined with lenalidomide/dexamethasone or pomalidomide/dexamethasone.12,13 Isatuximab is an immunoglobulin G1 (IgG1) monoclonal antibody that binds selectively to a specific epitope on the human CD38 receptor and is being evaluated in patients with MM. Isatuximab monotherapy was active and generally well tolerated in heavily pretreated patients with RRMM, with the greatest efficacy at doses ≥10 mg/kg (overall response rate [ORR], 24%).14 In a phase 1b study in heavily pretreated patients (95% had previously received lenalidomide and the majority were refractory to IMiDs), enhanced activity of isatuximab was seen in combination with lenalidomide/dexamethasone (ORR, 56%; n = 52).15
In addition, preclinical data have provided a strong rationale for combining isatuximab with pomalidomide or lenalidomide, as these agents significantly enhance the effector cell anti-MM cytotoxicity of isatuximab, inhibiting the growth of tumors.9,10 This phase 1b dose-escalation study (NCT02283775) evaluated the safety and tolerability of isatuximab in combination with standard doses of pomalidomide and low-dose dexamethasone in heavily pretreated patients with RRMM who had received prior treatment with lenalidomide and a PI.
Methods
Study population
Eligible patients were ≥18 years of age and had a documented diagnosis of MM according to International Myeloma Working Group (IMWG) criteria.16 Patients had received ≥2 previous therapies including lenalidomide and a PI, and had demonstrated disease progression during/after completion of their last therapy. Eligible patients had measurable disease: serum M protein ≥0.5 g/dL, urine M protein ≥200 mg/24 hours, or serum-free light chain ≥10 mg/dL and an abnormal serum-free light chain ratio (<0.26 or >1.65). Patients were required to have an absolute neutrophil count of ≥1000 cells per microliter (1.0 × 109/L) with no growth factor treatment in the previous 7 days, aspartate aminotransferase or alanine aminotransferase levels <2.5× upper limit of normal, platelet count ≥50 000 cells per microliter (50 × 109/L) (without platelet transfusion in the previous 7 days), total bilirubin ≤1.5× upper limit of normal, and calculated creatinine clearance ≥30 mL/min (Cockcroft-Gault equation).
Patients who had received prior therapy with pomalidomide were initially excluded from the study. However, to expand the patient population expected to benefit from the study treatment, the protocol was amended at the end of the 5 mg/kg dosing phase, allowing patients with previous pomalidomide exposure, and those who were not refractory to their last regimen, to enter the study.
Study design
This was a phase 1b, multicenter, open-label, noncomparative, dose-escalation study of isatuximab in combination with standard doses of pomalidomide/dexamethasone in patients with RRMM. After completion of the dose-escalation phase, additional patients were enrolled and treated at the recommended dose in an expansion cohort. A total of 6 sites in the United States participated in the study.
Patients received isatuximab 5, 10, or 20 mg/kg in 4 weekly doses (cycle 1), and every 2 weeks thereafter; pomalidomide 4 mg on days 1 to 21; and dexamethasone 40 mg (20 mg if aged ≥75 years) weekly in 28-day cycles until disease progression or intolerable toxicity. All patients received premedication consisting of IV/oral dexamethasone (administered as part of the premedication and the investigated combination treatment), diphenhydramine 25 to 50 mg IV (or equivalent), ranitidine 50 mg IV (or equivalent), and oral acetaminophen 650 to 1000 mg at least 15 to 30 minutes prior to each administration of isatuximab, to mitigate infusion-related reactions (IRRs). The initial infusion rate for isatuximab was 87.5 mg/h in the 5 mg/kg cohort, and 175.0 mg/h in both the 10 mg/kg and 20 mg/kg cohorts. The infusion rate was increased by 50 mg/h (first infusion) or 100 mg/h (subsequent infusions) every 30 minutes to a maximum of 400 mg/h. Primary prophylaxis with granulocyte–colony-stimulating factor (G-CSF) was permitted only after the dose-limiting toxicity (DLT) observation period (cycle 1).
Study end points and analyses
The primary objective was to evaluate safety and determine the recommended dose of isatuximab in combination with standard doses of pomalidomide/dexamethasone. The primary end point was the incidence of treatment-related DLTs during cycle 1, defined as grade 4 neutropenia lasting >7 days, grade 3/4 neutropenia complicated by fever or infection, grade 3/4 thrombocytopenia associated with bleeding requiring transfusion, or any grade ≥3 nonhematologic adverse event (AE) (excluding grade 3 fatigue, grade ≥3 electrolyte abnormalities, grade 3 nausea, vomiting, or diarrhea, or an allergic reaction to isatuximab). The safety of isatuximab in combination with pomalidomide/dexamethasone was evaluated using information from treatment-emergent AEs (TEAEs) and changes in laboratory parameters, vital signs, electrocardiograms, and physical examination. National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 was used to assess the severity of AEs.
The secondary end points included evaluation of efficacy (ORR, clinical benefit rate [CBR], progression-free survival [PFS], OS, and duration of response [DOR]), pharmacokinetic (PK) analysis, and immunogenicity of isatuximab and pomalidomide. ORR was defined as the proportion of patients with at least a partial response (PR) (IMWG criteria 2014).16 CBR was defined as the proportion of patients with at least a minimal response (IMWG criteria 2014).16 DOR was defined as the time from the date of the first response (PR or better) to the date of subsequent progressive disease or death, whichever occurred first. Clinical response was evaluated by M-protein quantification (serum and/or urine according to local laboratory results), serum-free light chain levels, bone marrow biopsy, or skeletal survey, and computed tomography or magnetic resonance imaging for the presence of plasmacytoma. For the PK analysis, plasma concentrations of isatuximab (all cycles with a limit of quantification of 0.5 ng/mL) and pomalidomide (cycles 1 and 3 with a limit of quantification of 0.22 ng/mL) were assayed using 2 validated methods: enzyme-linked immunosorbent assay for isatuximab and liquid chromatography/mass spectrometry for pomalidomide. PK assessment for both compounds was performed by noncompartmental analysis. Minimal residual disease (MRD) was evaluated as an exploratory end point in patients achieving at least a complete response (CR) by next-generation sequencing at a sensitivity of 10−6 in bone marrow samples at baseline and again at the time of CR (ImmunoSEQ pipeline by Adaptive Biotechnologies). Cytogenetic abnormalities (del17p, t[4:14], or t[4:16]) were also assessed at baseline (central laboratory).
Study oversight
The study protocol and subsequent amendments were approved by the ethics committee at every institution, and the study was conducted in accordance with recommendations of good clinical practice and the Declaration of Helsinki. All patients provided written informed consent before participating in the study. A study committee (investigator and sponsor) evaluated safety periodically to proceed with dose escalation.
Statistical analyses
The standard 3+3 design was used to evaluate escalating dose levels of isatuximab in combination with pomalidomide/dexamethasone. During the dose-escalation phase, a minimum of 3 patients were planned to be enrolled in each dose level and the actual sample size was determined by the number of dose levels evaluated and the emerging DLTs. After completion of the dose-escalation phase and determination of the recommended dose, 24 to 27 additional patients were to be enrolled at the selected dose (dose-expansion phase), to reach the 30 required patients to provide at least 80% power to reject the null hypothesis of true ORR of ≤30% when the true response rate was >50% based on a 1-sided exact binomial test at a significance level of 0.1. Safety and efficacy were evaluated in all patients who provided informed consent and who received at least 1 dose (even if incomplete) of isatuximab, pomalidomide, or dexamethasone (all-treated/safety population). The DLT-evaluable population included those patients from the dose-escalation phase of the trial who received the 4 planned doses of isatuximab and at least 75% of planned pomalidomide doses (16 of 21 doses) during cycle 1 (unless omitted due to a DLT), and with a DLT assessment at the end of cycle 1. PFS (time from the date of first study treatment administration to the date of first documentation of confirmed progressive disease, symptomatic deterioration, or death), DOR, and OS (time from the date of first study treatment administration to the date of death) were analyzed by the Kaplan-Meier method. Continuous data were summarized using descriptive statistics; categorical and ordinal data were summarized using number/percentage of patients.
Results
Patients
In total, 45 patients were enrolled between May 2015 and March 2017 and received isatuximab in combination with pomalidomide/dexamethasone. Of these, 23 patients were treated in the dose-escalation phase. Dose cohorts were enrolled sequentially to establish the maximum tolerated dose (MTD). In total, 8 patients were treated at a dose of 5 mg/kg isatuximab. One patient experienced a DLT (grade 4 neutropenia); therefore, 3 further patients were enrolled at this dose level. In addition, 2 patients who were not evaluable for DLTs were replaced. Six patients received isatuximab 10 mg/kg. One patient experienced a DLT (grade 4 neutropenic infection), resulting in the enrollment of 3 additional patients. No additional DLTs were reported in this cohort. A total of 6 patients received isatuximab 20 mg/kg. One patient experienced a DLT (grade 3 confusional state), and an additional 3 patients were enrolled. No additional DLTs were reported in this cohort. As the MTD was not reached in the dose-escalation cohort, with 1 DLT each at all dose levels (5 mg/kg, 10 mg/kg, and 20 mg/kg), the isatuximab recommended dose was chosen based on PK/pharmacodynamic modeling and simulations made for study TCD11863 (clinicaltrials.gov identifier: NCT01749969). Finally, 3 more patients were treated at the recommended dose of 10 mg/kg while waiting for the dose-expansion phase to open. A further 22 patients were treated at 10 mg/kg isatuximab in the dose-expansion phase leading to a total of 31 patients treated at this dose level.
The patient baseline characteristics are presented in Table 1. The median age was 67.0 years (range, 42-82 years). Most patients were heavily pretreated and had received a median of 5 prior regimens (range, 3-12) and 3 prior lines of therapy (range, 1-10), although 2 patients had received only 1 prior line of therapy (protocol deviation). All patients had been exposed to lenalidomide, of whom 82% were refractory. In total, 6 patients (13%) had received prior pomalidomide therapy, and 4 of these patients were refractory. Most patients were refractory to prior treatment with IMiDs (82.2%), PIs (84.4%), or both (71.1%). Approximately one-half of the patients were refractory to lenalidomide and bortezomib (51.1%), fewer than one-half were refractory to lenalidomide and carfilzomib (40.0%), and, to a lesser extent, refractory to lenalidomide, bortezomib, and carfilzomib (22.2%). In addition, 6 patients (13.3%) had high-risk cytogenetics (del17p [n = 4] or t[4:14] [n = 2]) at baseline. Some patients demonstrated grade 2 hematologic abnormalities at baseline: anemia (35.6%), leukopenia (20.0%), neutropenia (20.0%), lymphopenia (17.8%), and thrombocytopenia (17.8%).
Baseline characteristics and prior treatment
| Characteristic | Isatuximab dose in mg/kg and schedule | All patients, n = 45 | ||
|---|---|---|---|---|
| 5 QW/Q2W, n = 8 | 10 QW/Q2W,* n = 31 | 20 QW/Q2W, n = 6 | ||
| Median age (range), y | 65.0 (56-77) | 67.0 (42-82) | 65.5 (58-75) | 67.0 (42-82) |
| Median time since diagnosis (range), y | 3.46 (1.0-9.1) | 4.30 (1.6-15.8) | 5.18 (2.7-14.0) | 4.30 (1.0-15.8) |
| ISS stage at baseline, n (%) | ||||
| I | 5 (63) | 15 (48) | 2 (33) | 22 (49) |
| II | 2 (25) | 11 (36) | 4 (67) | 17 (38) |
| III | 1 (13) | 3 (10) | 0 | 4 (9) |
| Missing | 0 | 2 (7) | 0 | 2 (4) |
| Type of myeloma at diagnosis, n (%) | ||||
| IgA | 1 (13) | 4 (13) | 0 | 5 (11) |
| IgG | 3 (38) | 19 (61) | 5 (83) | 27 (60) |
| Light chain, κ and λ | 4 (50) | 7 (23) | 1 (17) | 12 (27) |
| Unknown | 0 | 1 (3) | 0 | 1 (2) |
| Cytogenetic risk at baseline, n (%) | ||||
| Standard | 6 (75) | 17 (55) | 3 (50) | 26 (58) |
| High* | 1 (13) | 3 (10) | 2 (33) | 6 (13) |
| Missing | 1 (13) | 11 (65) | 1 (17) | 13 (29) |
| Median bone marrow plasma cells at baseline, % (range) | 40 (5-90) | 25 (0-90) | 35 (11-80) | 27.8 (0-90) |
| Prior treatment of multiple myeloma and refractory status | ||||
| Median prior regimens, n (range) | 5 (3-7) | 5 (3-12) | 4 (4-5) | 5 (3-12) |
| Median prior lines, n (range) | 3 (2-4) | 3 (1-10) | 2 (1-4) | 3 (1-10) |
| Prior stem cell transplant, n (%) | ||||
| ≥1 | 6 (75) | 19 (61) | 5 (83) | 30 (67) |
| ≥2 | 2 (25) | 1 (3) | 2 (33) | 5 (11) |
| Refractory to, n (%) | ||||
| Lenalidomide | 6 (75) | 27 (87) | 4 (67) | 37 (82) |
| Pomalidomide | 0 | 3 (10) | 1 (17) | 4 (9) |
| Bortezomib | 5 (63) | 22 (71) | 2 (33) | 29 (64) |
| Carfilzomib | 3 (38) | 13 (42) | 2 (33) | 18 (40) |
| IMiD refractory, n (%) | 6 (75) | 27 (87) | 4 (67) | 37 (82) |
| PI refractory, n (%) | 7 (88) | 27 (87) | 4 (67) | 38 (84) |
| IMiD + PI refractory, n (%) | 5 (63) | 23 (74) | 4 (67) | 32 (71) |
| Refractory to last regimen, n (%) | 8 (100) | 29 (94) | 4 (67) | 41 (91) |
| Refractory to lenalidomide and bortezomib, n (%) | 3 (38) | 18 (58) | 2 (33) | 23 (51) |
| Refractory to lenalidomide and carfilzomib, n (%) | 3 (38) | 13 (42) | 2 (33) | 18 (40) |
| Refractory to lenalidomide, bortezomib, and carfilzomib, n (%) | 2 (25) | 8 (26) | 0 | 10 (22) |
| Refractory to lenalidomide, pomalidomide, bortezomib, and carfilzomib, n (%) | 0 | 2 (7) | 0 | 2 (4) |
| Characteristic | Isatuximab dose in mg/kg and schedule | All patients, n = 45 | ||
|---|---|---|---|---|
| 5 QW/Q2W, n = 8 | 10 QW/Q2W,* n = 31 | 20 QW/Q2W, n = 6 | ||
| Median age (range), y | 65.0 (56-77) | 67.0 (42-82) | 65.5 (58-75) | 67.0 (42-82) |
| Median time since diagnosis (range), y | 3.46 (1.0-9.1) | 4.30 (1.6-15.8) | 5.18 (2.7-14.0) | 4.30 (1.0-15.8) |
| ISS stage at baseline, n (%) | ||||
| I | 5 (63) | 15 (48) | 2 (33) | 22 (49) |
| II | 2 (25) | 11 (36) | 4 (67) | 17 (38) |
| III | 1 (13) | 3 (10) | 0 | 4 (9) |
| Missing | 0 | 2 (7) | 0 | 2 (4) |
| Type of myeloma at diagnosis, n (%) | ||||
| IgA | 1 (13) | 4 (13) | 0 | 5 (11) |
| IgG | 3 (38) | 19 (61) | 5 (83) | 27 (60) |
| Light chain, κ and λ | 4 (50) | 7 (23) | 1 (17) | 12 (27) |
| Unknown | 0 | 1 (3) | 0 | 1 (2) |
| Cytogenetic risk at baseline, n (%) | ||||
| Standard | 6 (75) | 17 (55) | 3 (50) | 26 (58) |
| High* | 1 (13) | 3 (10) | 2 (33) | 6 (13) |
| Missing | 1 (13) | 11 (65) | 1 (17) | 13 (29) |
| Median bone marrow plasma cells at baseline, % (range) | 40 (5-90) | 25 (0-90) | 35 (11-80) | 27.8 (0-90) |
| Prior treatment of multiple myeloma and refractory status | ||||
| Median prior regimens, n (range) | 5 (3-7) | 5 (3-12) | 4 (4-5) | 5 (3-12) |
| Median prior lines, n (range) | 3 (2-4) | 3 (1-10) | 2 (1-4) | 3 (1-10) |
| Prior stem cell transplant, n (%) | ||||
| ≥1 | 6 (75) | 19 (61) | 5 (83) | 30 (67) |
| ≥2 | 2 (25) | 1 (3) | 2 (33) | 5 (11) |
| Refractory to, n (%) | ||||
| Lenalidomide | 6 (75) | 27 (87) | 4 (67) | 37 (82) |
| Pomalidomide | 0 | 3 (10) | 1 (17) | 4 (9) |
| Bortezomib | 5 (63) | 22 (71) | 2 (33) | 29 (64) |
| Carfilzomib | 3 (38) | 13 (42) | 2 (33) | 18 (40) |
| IMiD refractory, n (%) | 6 (75) | 27 (87) | 4 (67) | 37 (82) |
| PI refractory, n (%) | 7 (88) | 27 (87) | 4 (67) | 38 (84) |
| IMiD + PI refractory, n (%) | 5 (63) | 23 (74) | 4 (67) | 32 (71) |
| Refractory to last regimen, n (%) | 8 (100) | 29 (94) | 4 (67) | 41 (91) |
| Refractory to lenalidomide and bortezomib, n (%) | 3 (38) | 18 (58) | 2 (33) | 23 (51) |
| Refractory to lenalidomide and carfilzomib, n (%) | 3 (38) | 13 (42) | 2 (33) | 18 (40) |
| Refractory to lenalidomide, bortezomib, and carfilzomib, n (%) | 2 (25) | 8 (26) | 0 | 10 (22) |
| Refractory to lenalidomide, pomalidomide, bortezomib, and carfilzomib, n (%) | 0 | 2 (7) | 0 | 2 (4) |
ISS, International Staging System; QW/Q2W, weekly administration in cycle 1 followed by every other week for subsequent cycles.
Duration of exposure was censored at day 1 in patients with no evaluable postbaseline disease assessment.
Of the 45 patients treated with isatuximab, 26 discontinued treatment due to disease progression (n = 18) as of the data cutoff of 8 May 2018, AEs (n = 2; grade 4 intestinal perforation and grade 3 IRR), or other reasons (n = 6), including unconfirmed disease progression (ie, absence of confirmatory assessment at the time of study treatment discontinuation; n = 3), patient/investigator decision (n = 2), and withdrawal of consent (n = 1). Patients received a median of 10 cycles of treatment (range, 1-29), and the median duration of exposure to all drugs was 9.6 months (0.2-26.2 months). The median relative dose intensities of isatuximab, pomalidomide, and dexamethasone were 93.4%, 69.9%, and 68.3%, respectively. Isatuximab dose omissions occurred in 22 patients (48.9%). Pomalidomide dose reductions and dexamethasone dose reductions occurred in 31 patients (68.9%) and 27 patients (60.0%), respectively.
Safety
One DLT was reported at each dose level: grade 4 neutropenia (5 mg/kg pomalidomide related), grade 4 neutropenic infection (10 mg/kg pomalidomide related), and grade 3 confusional state (20 mg/kg; isatuximab and pomalidomide related). None of these DLTs led to treatment discontinuation, and all resolved with dose omission of any drug or reduction of pomalidomide and/or dexamethasone. The MTD was not reached in the dose-escalation cohort. Overall, all patients experienced at least 1 TEAE, regardless of the relationship to study treatment, and 39 of 45 patients (86.7%) experienced a grade ≥3 TEAE. The most common TEAEs, excluding hematologic laboratory abnormalities and infusion reactions, were fatigue (62.2%), upper respiratory tract infections (URTIs) (42.2%), and dyspnea (40.0%) (Table 2). The most common grade ≥3 TEAE was pneumonia, which occurred in 8 patients (17.8%). The incidence of TEAEs did not appear to be dose dependent.
TEAEs and hematologic laboratory abnormalities
| TEAEs | Isatuximab dose in mg/kg and schedule | ||||
|---|---|---|---|---|---|
| No. of patients | All patients, n = 45 | ||||
| All grades/grade ≥3 | n (%) | ||||
| 5 QW/Q2W, n = 8 | 10 QW/Q2W, n = 31 | 20 QW/Q2W, n = 6 | All grades | Grade ≥3 | |
| Any TEAE | 8/8 | 31/26 | 6/5 | 45 (100) | 39 (87) |
| TEAE leading to dose reduction of pomalidomide | 5/4 | 25/21 | 4/4 | 34 (82) | 29 (64) |
| TEAE leading to dose reduction of dexamethasone | 3/1 | 27/13 | 4/3 | 34 (76) | 17 (38) |
| SAE | 5/5 | 17/16 | 4/4 | 26 (58) | 25 (56) |
| Fatigue | 5/1 | 18/1 | 5/1 | 28 (62) | 3 (7) |
| IRR | 4/0 | 14/1 | 1/0 | 19 (42) | 1 (2) |
| URTI | 4/0 | 12/0 | 3/0 | 19 (42) | 0 |
| Dyspnea | 5/1 | 11/1 | 2/1 | 18 (40) | 3 (7) |
| Constipation | 4/0 | 10/0 | 2/0 | 16 (36) | 0 |
| Diarrhea | 2/0 | 11/1 | 3/0 | 16 (36) | 1 (2) |
| Cough | 3/0 | 9/0 | 2/0 | 14 (31) | 0 |
| Headache | 4/0 | 8/0 | 2/0 | 14 (31) | 0 |
| Insomnia | 2/0 | 11/1 | 0/0 | 13 (29) | 1 (2) |
| Nausea | 2/0 | 7/0 | 3/0 | 12 (27) | 0 |
| Dizziness | 4/1 | 5/0 | 2/0 | 11 (24) | 1 (2) |
| Pyrexia | 2/0 | 5/1 | 3/0 | 10 (22) | 1 (2) |
| Pneumonia | 0/0 | 8/7 | 2/1 | 10 (22) | 8 (18) |
| Pain in extremity | 2/0 | 7/1 | 1/1 | 10 (22) | 2 (4) |
| Urinary tract infection | 1/0 | 6/2 | 1/1 | 8 (18) | 3 (7) |
| Hypertension | 2/1 | 4/2 | 1/0 | 7 (16) | 3 (7) |
| Traumatic fracture | 2/2 | 3/1 | 1/0 | 6 (13) | 3 (7) |
| Syncope | 1/1 | 2/2 | 0/0 | 3 (7) | 3 (7) |
| Hematologic laboratory abnormalities* | |||||
| Lymphopenia | 8/7 | 29/21 | 6/4 | 43 (98) | 32 (73) |
| Leukopenia | 8/7 | 29/19 | 6/5 | 43 (98) | 31 (71) |
| Anemia | 8/0 | 29/3 | 6/2 | 43 (98) | 5 (11) |
| Neutropenia | 8/7 | 27/24 | 6/6 | 41 (93) | 37 (84) |
| Thrombocytopenia | 7/2 | 25/9 | 5/4 | 37 (84) | 15 (34) |
| TEAEs | Isatuximab dose in mg/kg and schedule | ||||
|---|---|---|---|---|---|
| No. of patients | All patients, n = 45 | ||||
| All grades/grade ≥3 | n (%) | ||||
| 5 QW/Q2W, n = 8 | 10 QW/Q2W, n = 31 | 20 QW/Q2W, n = 6 | All grades | Grade ≥3 | |
| Any TEAE | 8/8 | 31/26 | 6/5 | 45 (100) | 39 (87) |
| TEAE leading to dose reduction of pomalidomide | 5/4 | 25/21 | 4/4 | 34 (82) | 29 (64) |
| TEAE leading to dose reduction of dexamethasone | 3/1 | 27/13 | 4/3 | 34 (76) | 17 (38) |
| SAE | 5/5 | 17/16 | 4/4 | 26 (58) | 25 (56) |
| Fatigue | 5/1 | 18/1 | 5/1 | 28 (62) | 3 (7) |
| IRR | 4/0 | 14/1 | 1/0 | 19 (42) | 1 (2) |
| URTI | 4/0 | 12/0 | 3/0 | 19 (42) | 0 |
| Dyspnea | 5/1 | 11/1 | 2/1 | 18 (40) | 3 (7) |
| Constipation | 4/0 | 10/0 | 2/0 | 16 (36) | 0 |
| Diarrhea | 2/0 | 11/1 | 3/0 | 16 (36) | 1 (2) |
| Cough | 3/0 | 9/0 | 2/0 | 14 (31) | 0 |
| Headache | 4/0 | 8/0 | 2/0 | 14 (31) | 0 |
| Insomnia | 2/0 | 11/1 | 0/0 | 13 (29) | 1 (2) |
| Nausea | 2/0 | 7/0 | 3/0 | 12 (27) | 0 |
| Dizziness | 4/1 | 5/0 | 2/0 | 11 (24) | 1 (2) |
| Pyrexia | 2/0 | 5/1 | 3/0 | 10 (22) | 1 (2) |
| Pneumonia | 0/0 | 8/7 | 2/1 | 10 (22) | 8 (18) |
| Pain in extremity | 2/0 | 7/1 | 1/1 | 10 (22) | 2 (4) |
| Urinary tract infection | 1/0 | 6/2 | 1/1 | 8 (18) | 3 (7) |
| Hypertension | 2/1 | 4/2 | 1/0 | 7 (16) | 3 (7) |
| Traumatic fracture | 2/2 | 3/1 | 1/0 | 6 (13) | 3 (7) |
| Syncope | 1/1 | 2/2 | 0/0 | 3 (7) | 3 (7) |
| Hematologic laboratory abnormalities* | |||||
| Lymphopenia | 8/7 | 29/21 | 6/4 | 43 (98) | 32 (73) |
| Leukopenia | 8/7 | 29/19 | 6/5 | 43 (98) | 31 (71) |
| Anemia | 8/0 | 29/3 | 6/2 | 43 (98) | 5 (11) |
| Neutropenia | 8/7 | 27/24 | 6/6 | 41 (93) | 37 (84) |
| Thrombocytopenia | 7/2 | 25/9 | 5/4 | 37 (84) | 15 (34) |
TEAEs were defined as AEs that developed, worsened, or became serious during the TEAE period. The TEAE period was defined as the time from first dose of isatuximab, pomalidomide, or dexamethasone, whichever occurred first, up to 30 days after the last dose.
SAE, serious adverse event. See Table 1 for expansion of other abbreviations.
n = 30 for 10 mg/kg cohort; n = 44 for all patients.
In total, IRRs were reported in 19 patients (42.2%), including 1 grade 3 IRR in a patient receiving 10 mg/kg, which resulted in definitive study treatment discontinuation, per the protocol. All other IRRs were grade 1/2 in severity. The majority of IRRs occurred during the first infusion (18 patients; 42.2%), with IRRs occurring at later infusions in 3 patients (6.7%) (Figure 1). Neither the incidence nor severity of IRRs appeared to be dependent on isatuximab dose. The infusion rate was increased following the first infusion, thus allowing the infusion time to be reduced (median infusion time at 10 mg/kg dose was 3.3 hours for the first infusion and 2.9 hours for subsequent infusions).
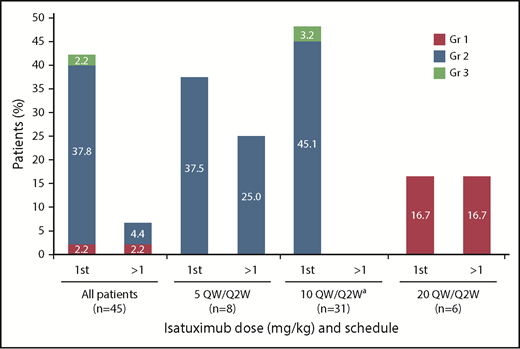
Infusion reactions by infusion and dose.aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. Gr, grade; QW/Q2W, weekly administration in cycle 1 followed by every other week for subsequent cycles.
Infusion reactions by infusion and dose.aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. Gr, grade; QW/Q2W, weekly administration in cycle 1 followed by every other week for subsequent cycles.
Hematologic laboratory abnormalities (grade 3/4) were common: neutropenia, 84.4%; lymphopenia, 72.7%; leukopenia, 70.4%; thrombocytopenia, 34.1%; and anemia, 11.4%. The most common grade ≥3 laboratory abnormality was neutropenia (grade 3, 26.7%; grade 4, 55.6%). All cases of neutropenia were manageable with dose modification (22 patients [48.9%]) and/or G-CSF support (26 patients [57.8%]). The median time from first dose to onset of first grade ≥3 neutropenia was 22 days. Ten patients (22.2%), 37 patients (82.2%), 27 patients (60.0%), and 17 patients (37.8%) had 1, ≥1, ≥2, or ≥3 episodes of grade ≥3 laboratory neutropenia, respectively. Neutropenic complications consisted of grade 4 neutropenic infection in 1 patient. There were no reports of febrile neutropenia and no patients withdrew from the trial due to neutropenia. The median duration of grade ≥3 neutropenia was 9 days (95% confidence interval [CI], 8-14), and the incidence decreased following the use of G-CSF and dose modification.
Other common grade ≥3 hematologic laboratory abnormalities included lymphopenia (72.7%) and leukopenia (70.4%); transfusions of packed red blood cells and platelets were performed in 10 patients (22.2%) and 3 patients (6.7%), respectively. No transfusion-related hemolysis occurred during the study.
One grade 4 neutropenic infection (2.2%) was reported, along with other grade ≥3 infections such as pneumonia in 8 patients (17.8%) and urinary tract infections in 3 patients (6.7%). Dose reduction of both pomalidomide and dexamethasone due to AEs occurred in 34 patients (75.6%). TEAEs (all grades) leading to pomalidomide dose reduction in >10% of patients were neutropenia (46.7%), URTI (17.8%), pneumonia (13.3%), and fatigue (11.1%). TEAEs leading to dexamethasone dose reduction in >10% of patients were insomnia (20.0%), pneumonia, fatigue, neutropenia (15.6% each), and URTI (13.3%). Two patients permanently discontinued all treatment due to TEAEs, including 1 patient with an intestinal perforation due to underlying MM, which led to death (not related to isatuximab) and 1 patient with a grade 3 IRR. Five patients (11.1%) died within 30 days of their last study treatment administration: 1 due to an AE (intestinal perforation) and 4 due to progressive disease. Five additional patients died during the posttreatment period due to progressive disease.
Pharmacokinetics
Isatuximab PK exposure parameters after the first administration are described in Table 3. Plasma concentrations of isatuximab and pomalidomide were measured in all patients treated at the various dose levels (isatuximab: 5, 10, and 20 mg/kg; pomalidomide: 2, 3, and 4 mg). Isatuximab (area under curve [AUC]1week) increased in a greater than dose-proportional manner over the dose range of 5 to 20 mg/kg weekly/every 2 weeks. However, no major deviation from dose proportionality was observed between 10 and 20 mg/kg, with a 2.2-fold increase in exposure for a 2-fold increase in dose. The accumulation at cycle 3 (seventh administration) compared with first administration was ∼2.3. Pomalidomide PK parameters at the 4-mg dose were determined for all patients after the first administration in cycles 1 and 3. PK parameters of pomalidomide were comparable after repeated administrations compared with those after the first pomalidomide dose, with no accumulation observed (AUC0-24 585 μg·h/mL [coefficient of variation (CV), 21.6%]).
Isatuximab PK parameters
| Isatuximab (plasma) | |||
|---|---|---|---|
| 5 mg/kg QW/Q2W | 10 mg/kg QW/Q2W | 20 mg/kg QW/Q2W | |
| Cycle 1 | |||
| n | 5 | 18 | 6 |
| Cmax, mean ± SD (geometric mean) [CV%], µg/mL | 91.3 ± 19.8 (89.7) [21.7] | 141 ± 18.8 (140) [13.3] | 297 ± 16.7 (296) [5.6] |
| tmax, median (min-max), h | 2.42 (2.13-3.83) | 3.41 (2.30-28.00) | 5.22 (4.28-8.93) |
| AUC1week, mean ± SD (geometric mean) [CV%], μg·h/mL | 6100 ± 2840 (5580) [46.5] | 12 800 ± 2430* (12 600) [18.9] | 27 000 ± 5620 (26 500) [20.8] |
| Cycle 3 | |||
| n | 6 | 24 | 6 |
| Cmax, mean ± SD (geometric mean) [CV%], µg/mL | 167 ± 34.5 (164) [20.6] | 403 ± 163 (375) [40.4] | 648 ± 246 (611) [38.0] |
| tmax, median (min-max), h | 5.97 (1.83-7.25) | 6.36 (2.00-23.67) | 7.52 (4.25-48.17) |
| AUC2weeks, mean ± SD (geometric mean) [CV%], μg·h/mL | 30 900 ± 10 900 (29 000) [35.2] | 71 000 ± 34 600† (63 990) [48.7] | 156 000 ± 91 000‡ (134 000) [58.5] |
| Isatuximab (plasma) | |||
|---|---|---|---|
| 5 mg/kg QW/Q2W | 10 mg/kg QW/Q2W | 20 mg/kg QW/Q2W | |
| Cycle 1 | |||
| n | 5 | 18 | 6 |
| Cmax, mean ± SD (geometric mean) [CV%], µg/mL | 91.3 ± 19.8 (89.7) [21.7] | 141 ± 18.8 (140) [13.3] | 297 ± 16.7 (296) [5.6] |
| tmax, median (min-max), h | 2.42 (2.13-3.83) | 3.41 (2.30-28.00) | 5.22 (4.28-8.93) |
| AUC1week, mean ± SD (geometric mean) [CV%], μg·h/mL | 6100 ± 2840 (5580) [46.5] | 12 800 ± 2430* (12 600) [18.9] | 27 000 ± 5620 (26 500) [20.8] |
| Cycle 3 | |||
| n | 6 | 24 | 6 |
| Cmax, mean ± SD (geometric mean) [CV%], µg/mL | 167 ± 34.5 (164) [20.6] | 403 ± 163 (375) [40.4] | 648 ± 246 (611) [38.0] |
| tmax, median (min-max), h | 5.97 (1.83-7.25) | 6.36 (2.00-23.67) | 7.52 (4.25-48.17) |
| AUC2weeks, mean ± SD (geometric mean) [CV%], μg·h/mL | 30 900 ± 10 900 (29 000) [35.2] | 71 000 ± 34 600† (63 990) [48.7] | 156 000 ± 91 000‡ (134 000) [58.5] |
Cmax, peak serum concentration; SD, standard deviation; tmax, time when peak serum concentration is reached. See Table 1 for expansion of other abbreviations.
n = 16.
n = 19.
n = 5.
Efficacy
At a median overall follow-up duration of 8.6 months (0-25.8 months) across all dose groups, ORR was 62.2%, including 16 PRs, 10 very good partial responses (VGPRs), 1 CR, and 1 stringent complete response (sCR) (Figure 2A). The ORR in patients receiving isatuximab 10 mg/kg (n = 31) was 64.5%, including 13 PRs, 6 VGPRs, and 1 sCR. The CBR was 73.3%. Disease progression was reported as the best response in only 3 patients (6.7%), including 1 patient in the 5 mg/kg and 2 patients in 10 mg/kg cohorts. Two patients in the all-treated population achieved at least a CR and were assessed for MRD: 1 patient 5 months after initiation of study treatment and the other 7.5 months after initiation of study treatment; both were MRD+. Among 37 IMiD-refractory patients treated at any dose level, the ORR was 56.8%. The ORR in PI-refractory patients (n = 38) was 63.2%, and in IMiD- and PI-refractory patients (n = 32) the best ORR was 59.4%. The ORR in lenalidomide- and bortezomib-refractory patients (n = 23) was 65.2%, and in lenalidomide- and carfilzomib-refractory patients (n = 18) was 50%. The ORR in patients refractory to lenalidomide, bortezomib, and carfilzomib (n = 10) was 60.0% (Figure 2B). Among the 4 patients who were refractory to pomalidomide, 1 patient had a PR or better.
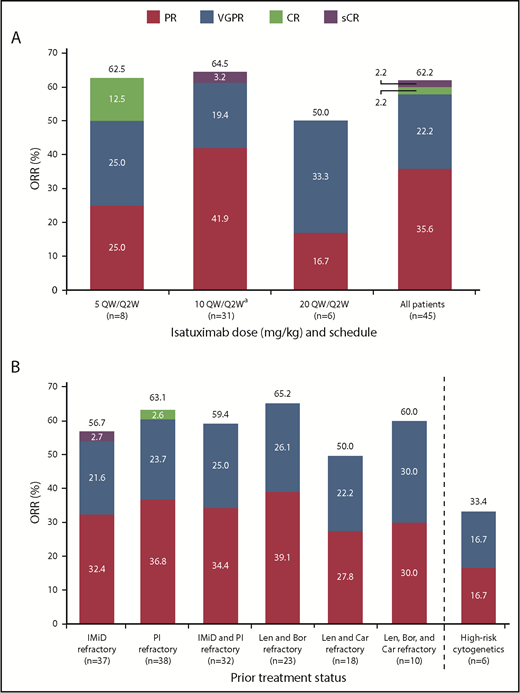
Confirmed clinical response. (A) ORR for all patients and by dose. (B) ORR for subgroups. aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. Bor, bortezomib; Car, carfilzomib; Len, lenalidomide; sCR, stringent complete response; VGPR, very good partial response.
Confirmed clinical response. (A) ORR for all patients and by dose. (B) ORR for subgroups. aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. Bor, bortezomib; Car, carfilzomib; Len, lenalidomide; sCR, stringent complete response; VGPR, very good partial response.
The responses were durable with a median DOR of 18.7 months (95% CI, 12.5 months to not reached [NR]). In addition, patients responded quickly, with a median time to first response of 0.9 months (range, 0.9-5.1 months) (Figure 3). Median PFS was 17.6 months (95% CI, 6.8-20.5 months) in the all-treated population, and 17.6 months (6.80 months to NR) in the 10 mg/kg dose group (Figure 4). Median OS was not reached (95% CI, 17.58 to NR).
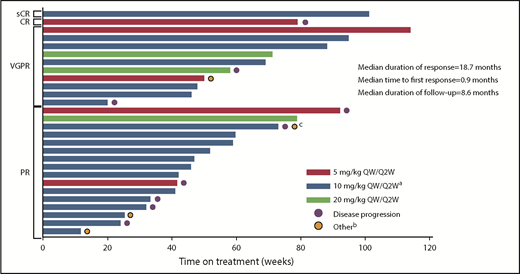
Time on treatment by best confirmed response (at least PR).aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. bOther reasons for discontinuation were unconfirmed disease progression (n = 3) and withdrawal of consent (n = 1). cPatient had progressive disease and withdrew consent.
Time on treatment by best confirmed response (at least PR).aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. bOther reasons for discontinuation were unconfirmed disease progression (n = 3) and withdrawal of consent (n = 1). cPatient had progressive disease and withdrew consent.
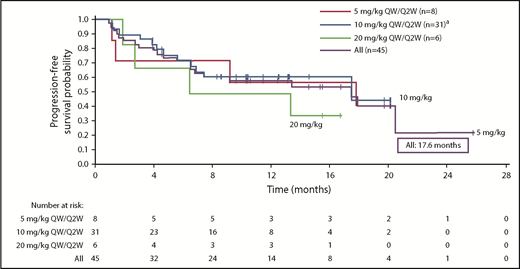
Kaplan-Meier analysis of PFS.aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. Median PFS (95% CI) is reported for each dose cohort.
Kaplan-Meier analysis of PFS.aIndicates data representing dose-escalation cohort (n = 9) and expansion cohort (n = 22) combined. Median PFS (95% CI) is reported for each dose cohort.
Discussion
This was a phase 1b dose-escalation study to evaluate the safety and preliminary efficacy of isatuximab in combination with standard doses of pomalidomide/dexamethasone. Overall, this combination was well tolerated up to the highest planned dose (20 mg/kg) of isatuximab. DLTs were observed in 3 patients, 1 at each dose level, and proved manageable with dose modification of pomalidomide or omission of isatuximab and/or pomalidomide.
Although cross-trial comparisons should be interpreted with caution, the safety profile of isatuximab in combination with pomalidomide/dexamethasone appears to be consistent with the safety profile of the individual drugs,17 with the exception of neutropenia. Despite the high incidence of serious TEAEs with grade ≥3 severity (55.6%), and high-dose reductions of pomalidomide (75.6%) and dexamethasone (75.6%) caused by TEAEs, definitive treatment discontinuation due to AEs was not very common (4.4%). TEAEs leading to dose reduction of pomalidomide occurred in 75.6% of patients; however, these were manageable. The high incidence of overall dose reductions for pomalidomide (68.9%) and dexamethasone (60.0%) did not impact the efficacy outcomes of the study.
Overall, IRRs occurred in fewer than one-half of patients (42.2%), with most events occurring during the first infusion. With the exception of 1 patient with a grade 3 IRR, these were all grade 1/2. By comparison, the incidence of IRRs observed was 48.9% with isatuximab monotherapy,14 55.6% with isatuximab in combination with lenalidomide/dexamethasone,15 and 50% with daratumumab in combination with pomalidomide/dexamethasone.13 Because investigators are gaining experience with monoclonal antibodies, including anti-CD38, they are better able to manage IRRs compared with earlier trials. In addition, the infusion rate for isatuximab could be increased after the first infusion, and prophylaxis could be reconsidered based on patient tolerability, at the investigator’s discretion. The median infusion time for isatuximab at the 10 mg/kg dose was 3.3 hours for the first infusion and 2.9 hours for subsequent infusions. By comparison, daratumumab requires a long infusion time (8.2 hours for the initial infusion, 6.5 hours for the second infusion, and 3.9 hours for subsequent infusions) and prophylaxis pre- and postinfusion.12,13,18,19
The incidence of grade ≥3 neutropenia was higher in this study (84.1%) than with either isatuximab as a single agent (14.9%)14 or pomalidomide/dexamethasone alone (47.7%).20 However, there were no cases of febrile neutropenia; grade 4 neutropenic infection was reported in 1 patient, and no patients withdrew from the study due to neutropenia. All cases of grade ≥3 neutropenia were manageable with dose modifications (dose reduction of pomalidomide or dose delay/omission of isatuximab and/or pomalidomide) and/or the use of G-CSF. The preexisting incidence of grade 1/2 neutropenia at baseline (22.2%) and the lack of primary G-CSF prophylaxis during cycle 1 may also have contributed to the high incidence of grade 3/4 neutropenia. The median time to first onset of grade ≥3 neutropenia was 22 days, which explains the early dose reductions of pomalidomide.
After the first administration of isatuximab 10 mg/kg in combination with pomalidomide/dexamethasone, isatuximab exposure (AUC1week, 12 800 µg·h/mL; CV, 24.3%) was comparable to that observed with isatuximab monotherapy (mean AUC1week, 17 000 μg·h/mL; CV, 22%).14 The PK parameters of isatuximab were unaffected by coadministration with pomalidomide/dexamethasone. Pomalidomide exposure (AUC0-24h, 585 μg·h/mL; CV, 21.6%) was comparable to the exposure previously reported for pomalidomide in combination with dexamethasone (638 μg·h/mL; CV, 43.0).21 These results suggest that there is no interaction between isatuximab and pomalidomide when given in combination.
Patients in the current study had been heavily pretreated, with a median of 3 prior lines of therapy; some patients had received up to 10 prior lines of therapy. Additionally, most were refractory to the current standard of care therapies (lenalidomide, 82.2%; PIs, 84.4%; and IMiDs and PIs, 71.1%). The ORR in the all-treated population in this study was 62.2%, which is twofold higher than historical observations with pomalidomide/dexamethasone alone (28% to 31%).22,23 Similarly, the VGPR and CR rates were higher in this study compared with pomalidomide/dexamethasone alone (VGPR, 22.2% vs 7.6%; CR, 2.2% vs 0.6%). The VGPR and CR rates may be underestimated here due to monoclonal antibody interference in serum immunofixation electrophoresis to detect M protein. An assay to assess this potential interference for isatuximab is being developed.
Median DOR, PFS, and OS data appear very promising; however, these data are still maturing, with a median follow-up of 8.6 months, with 42.2% of patients still receiving treatment, and with a small sample size. In addition, the median DOR in the current study was higher compared with previous observations for pomalidomide/dexamethasone alone (18.7 vs 7.0 months, respectively).22 The ORR in this study was 62.2% and the OS at 12 months was 80%. Moreover, these results are comparable to those observed in 2 previous phase 1b studies of daratumumab in combination with pomalidomide/dexamethasone: ORR of 66% and 60% and OS at 12 months of 52% and 89%, respectively.13,24 The ORR in this study also compares favorably with results from a study in which elotuzumab in combination with pomalidomide/dexamethasone demonstrated an ORR of 53%, whereas the OS was not mature at the time of publication.25
In conclusion, the results of this phase 1b study demonstrate that isatuximab in combination with pomalidomide/dexamethasone is well tolerated with promising activity in heavily pretreated patients with RRMM. Based on efficacy, safety, and PK data, the 10 mg/kg dose of isatuximab was selected for the expansion cohort and for future combination studies. The efficacy and safety observed in this heavily pretreated population demonstrate that isatuximab may be an effective treatment option for patients with a high burden of disease who have become refractory to lenalidomide and PIs. A new cohort for this study (Part B) will explore the incidence and duration of grade ≥3 IRRs from a fixed volume of isatuximab infusion. A global phase 3 study (ICARIA; NCT02990338) comparing safety and efficacy of isatuximab 10 mg/kg weekly/every 2 weeks in combination with pomalidomide/dexamethasone vs pomalidomide/dexamethasone alone is ongoing.26
Qualified researchers may request access to patient-level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and data-set specifications. Patient-level data will be anonymized, and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data-sharing criteria, eligible studies, and process for requesting access can be found at: https://www.clinicalstudydatarequest.com/.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the participating patients and their families, the study centers, and the investigators for their contributions to the study, and the following individuals from Sanofi for their respective contributions: Olivier Vitse and Samira Ziti-Ljajic (PK analyses); Lei Gao, Ye Zhou, Pamela Butler, and Jinrong Hou (statistical support); and Sandrine Macé (examination of exploratory end points). Editorial assistance was provided by Louise Wright of Adelphi Communications Ltd (funded by Sanofi).
This work was supported by Sanofi.
Sanofi collaborated with the authors on the designing of the study as well as the collection, analysis, and interpretation of data.
Authorship
Contribution: J.M., P.R., and F.C. conceived and designed the study; J.M., P.R., S.Z.U., N.R., W.B., C.K., and K.A. provided study material or patients and collected and/or assembled data; J.M., P.R., F.C., D.K., F.D., Q.L., and D.S. analyzed and interpreted data; and J.M., P.R., S.Z.U., N.R., W.B., C.K., F.C., D.K., F.D., Q.L., D.S., and K.A. wrote, and gave final approval of, the manuscript.
Conflict-of-interest disclosure: J.M. received institutional research funding from AbbVie, Celgene, Onyx, and Sanofi. P.R. has a consulting or advisory role with Genmab, and has received research funding from Celgene and Millennium. S.Z.U. has received honoraria from Celgene, Skyline, Onyx, Millennium, Sanofi, and Janssen; has a consulting or advisory role with Amgen, Bristol-Myers Squibb, Celgene, Janssen, Merck, SkylineDx, and Takeda; has been on speakers’ bureaus for Amgen, Celgene, Janssen, Sanofi, and Takeda; and has received research funding from Amgen, Array Biopharma, Bristol-Myers Squibb, Celgene, Janssen, Merck, Pharmacyclics, Sanofi, and Takeda. N.R. has a consulting or advisory role with Amgen, Novartis, Takeda, and Celgene, and has received research funding from AstraZeneca. W.B. has received honoraria from Amgen, Celgene, and Acetylon; has a consulting or advisory role with Amgen, Celgene, Sanofi, Merck, and Bristol-Myers Squibb; has been on speakers’ bureaus for Celgene, Amgen, and Takeda; has received research funding from Acetylon, Bristol-Myers Squibb, Celgene, Karyopharm Therapeutics, Merck, Amgen, and Sanofi; and has given expert testimony on behalf of Takeda and Celgene. C.K. is an employee of City of Hope, and has received research funding from Sanofi, Pharmacyclics, and Celgene. F.C., D.K., F.D., Q.L., and D.S. are employees of, and hold stock or other ownership interests in, Sanofi. K.A. holds stock or other ownership interests in, and is scientific founder of, Oncopep C4 Therapeutics, and has a consulting or advisory role with Millennium, Takeda, Gilead, Bristol-Myers Squibb, Celgene, and Janssen.
Correspondence: Joseph Mikhael, City of Hope Cancer Center and International Myeloma Foundation, 445 N Fifth St, Phoenix, AZ 85004; e-mail: jmikhael@myeloma.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal