

Key Points
PF4iCre;JAK2V617F/WT mice develop a full MPN that mimics polycythemia vera.
The PF4iCre system induces JAK2V617F mutation in a small subset of HSC.

Abstract
The major weakness of most knock-in JAK2V617F mouse models is the presence of the JAK2 mutation in all rather than in a few hematopoietic stem cells (HSC), such as in human “early-stage” myeloproliferative neoplasms (MPN). Understanding the mechanisms of disease initiation is critical as underscored by the incidence of clonal hematopoiesis of indeterminate potential associated with JAK2V617F. Currently, such studies require competitive transplantation. Here, we report a mouse model obtained by crossing JAK2V617F/WT knock-in mice with PF4iCre transgenic mice. As expected, PF4iCre;JAK2V617F/WT mice developed an early thrombocytosis resulting from the expression of JAK2V617F in the megakaryocytes. However, these mice then developed a polycythemia vera–like phenotype at 10 weeks of age. Using mT/mG reporter mice, we demonstrated that Cre recombination was present in all hematopoietic compartments, including in a low number of HSC. The frequency of mutated cells increased along hematopoietic differentiation mimicking the clonal expansion observed in essential thrombocythemia and polycythemia vera patients. This model thus mimics the HSC compartment observed in early-stage MPN, with a small number of JAK2V617F HSC competing with a majority of JAK2WT HSC. PF4iCre;JAK2V617F/WT mice are a promising tool to investigate the mechanisms that regulate clonal dominance and progression to myelofibrosis.
Introduction
Myeloproliferative neoplasms (MPN) associated with JAK2V617F mutation have been modeled through different means, among others, knock-in (KI) models in which JAK2V617F is expressed under the control of its own endogenous promoter. This can be achieved using the Cre-Lox technology based on a tissue-specific expression of the Cre-recombinase. However, in previous KI models (Tie2-Cre, Vav-Cre, E2A-Cre),1-3 the JAK2V617F mutation has been introduced in nearly all hematopoietic stem cells (HSC), in contrast to essential thrombocythemia (ET) and polycythemia vera (PV) patients, who only carry a minority of mutated HSC.4 Because JAK2V617F in HSC has a low fitness to give rise to a disease,5-8 assessing the mechanisms ruling JAK2V617F clonal dominance is crucial but requires a mouse model that mimics the HSC compartment of ET and PV patients. Here, we observed that the PF4iCre system allows expression of JAK2V617F in a minority of HSC leading to a model in which a small subset of JAK2V617F HSC induces a full-blown MPN phenotype.
Methods
Generation of mouse models
PF4iCre;JAK2V617F/WT mice were generated by crossing JAK2V617F KI mice9 with PF4iCre transgenic mice.10 PF4iCre; mTmG;JAK2V617F/WT mice were then generated by crossing PF4iCre;JAK2V617F/WT with ROSAmTmG mice.11 PdgfbiCre-ERT2;JAK2V617F/WT were previously described.12 This study was conducted in accordance with both Bordeaux University Institutional Committee guidelines (committee CEEA50) and those in force in the European Community for experimental animal use (L358-86/609/EEC).
Experimental methodologies
All methods are detailed as outlined in the supplemental Methods, available on the Blood Web site.
Results and discussion
PF4iCre;JAK2V617F/WT mice develop a typical JAK2V617F clonal MPN
Because different groups reported recombination in a few subsets of HSC in the PF4iCre mouse model,10,13,14 we developed a conditional JAK2V617F-driven MPN mouse model by crossing conditional heterozygous JAK2V617F/WT KI mice9 with PF4iCre mice. As expected, we observed an early thrombocytosis in PF4iCre;JAK2V617F/WT mice mimicking an ET (Figure 1A). A few weeks after, the hematocrit and the leukocyte counts increased, indicative of an evolution toward a PV-like disease (Figure 1A). PF4iCre;JAK2V617F/WT mice developed a splenomegaly (Figure 1B) with red pulp hyperplasia compatible with a PV phenotype (Figure 1C). There was no evidence of fibrosis, consistent with the absence of cytopenia. The clonal origin of this MPN phenotype was demonstrated by the expression of JAK2V617F in all myeloid lineages, with a variant allele frequency of ∼10% in erythroid cells, 9% in granulocytes, and 50% in megakaryocytes (MK) (Figure 1D-E). This corresponds to a frequency of JAK2 mutated cells of 20%, 18%, and 100%, respectively. The mutation remained undetectable in control mice (Figure 1D-E).
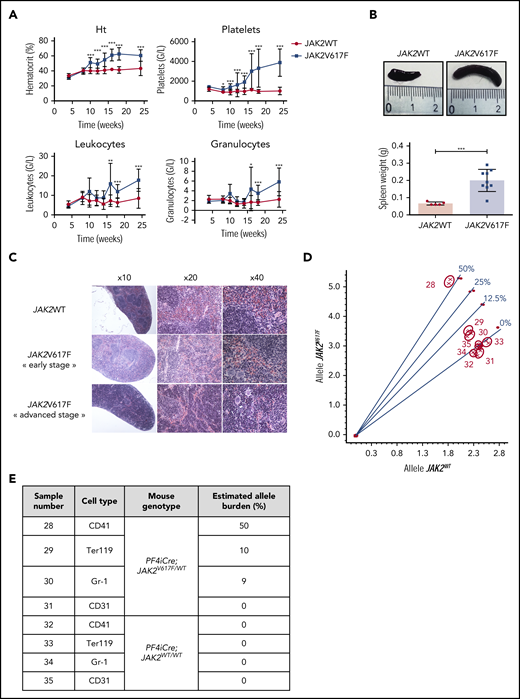
PF4iCre;JAK2V617F/WTmice develop a typical MPN with detection of JAK2V617F mutation in myeloid cells. (A) Blood cell counts of PF4-iCre;JAK2V617F/WT mice and littermate controls. Data are presented as mean ± standard deviation (n = 5-24 JAK2WT; n = 5-29 JAK2V617F). Statistical significance determined by 2-tailed unpaired t test. *P < .05; **P < .01; ***P < .001; (B) Spleen weight (also showing spleens collected) in 12-week-old PF4iCre;JAK2V617F/WT mice. Data are presented as individual values and mean ± standard error of the mean (n = 5 JAK2WT; n = 9 JAK2V617F). (C) Histologic examination of the spleen (HES coloration). (Top) Representative normal splenic architecture observed in 12-week-old PF4iCre;JAK2WT/WT mice. (Middle) Histological features observed in 12-week-old PF4iCre;JAK2V617F/WT mice with myeloid hyperplasia without disturbance of the white pulp. (Bottom) Histological features observed in 18-week-old PF4iCre;JAK2V617F/WT mice with myeloid hyperplasia associated with a near disappearance of the white pulp. (D) Bone marrow myeloid cells from 12-week-old were sorted based on the expression of CD42 (megakaryocytes), Gr-1 (Ly6G+C, granulocytes precursors), Ter119 (erythroid cells), or CD31 (endothelial cells); the JAK2V617F mutation was searched in these cell populations by allele-specific genotyping. The graph represents the wild-type allele relative quantity on the x-axis and the V617F allele relative quantity on the y-axis. Standard curves (0%, 12.5%, 25%, 50%) of V617F allele burden are indicated by circles. Samples are indicated by crosses. (E) The table details the results of allelic burdens observed for the different cell populations sorted in wild-type and mutated mice, as determined from the discrimination plot (D).
PF4iCre;JAK2V617F/WTmice develop a typical MPN with detection of JAK2V617F mutation in myeloid cells. (A) Blood cell counts of PF4-iCre;JAK2V617F/WT mice and littermate controls. Data are presented as mean ± standard deviation (n = 5-24 JAK2WT; n = 5-29 JAK2V617F). Statistical significance determined by 2-tailed unpaired t test. *P < .05; **P < .01; ***P < .001; (B) Spleen weight (also showing spleens collected) in 12-week-old PF4iCre;JAK2V617F/WT mice. Data are presented as individual values and mean ± standard error of the mean (n = 5 JAK2WT; n = 9 JAK2V617F). (C) Histologic examination of the spleen (HES coloration). (Top) Representative normal splenic architecture observed in 12-week-old PF4iCre;JAK2WT/WT mice. (Middle) Histological features observed in 12-week-old PF4iCre;JAK2V617F/WT mice with myeloid hyperplasia without disturbance of the white pulp. (Bottom) Histological features observed in 18-week-old PF4iCre;JAK2V617F/WT mice with myeloid hyperplasia associated with a near disappearance of the white pulp. (D) Bone marrow myeloid cells from 12-week-old were sorted based on the expression of CD42 (megakaryocytes), Gr-1 (Ly6G+C, granulocytes precursors), Ter119 (erythroid cells), or CD31 (endothelial cells); the JAK2V617F mutation was searched in these cell populations by allele-specific genotyping. The graph represents the wild-type allele relative quantity on the x-axis and the V617F allele relative quantity on the y-axis. Standard curves (0%, 12.5%, 25%, 50%) of V617F allele burden are indicated by circles. Samples are indicated by crosses. (E) The table details the results of allelic burdens observed for the different cell populations sorted in wild-type and mutated mice, as determined from the discrimination plot (D).
JAK2V617F knock-in is not restricted to MK in PF4iCre;JAK2V617F/WT mice
To precisely measure the size of the JAK2V617F clone, we crossed PF4iCre;JAK2V617F/WT mice with reporter mT/mG mice.11 PF4iCre;mT/mG;JAK2V617F/WT mice developed a PV-like phenotype with similar kinetics as PF4iCre;JAK2V617F/WT mice, but with a more limited thrombocytosis (supplemental Figure 1). This difference previously reported1,15,16 highlights the effect of the genetic background on the phenotypic expression of an oncogenic mutation.17,18 As expected, GFP expression was observed in nearly all platelets of control and JAK2-mutated mice (Figure 2A). However, GFP was also detectable in other myeloid lineages at a higher level in PF4iCre;mT/mG;JAK2V617F/WT than in control mice (16.6 ± 3.6% vs 0.52 ± 0.09% for Ter119+ cells, P < .001, 16.0 ± 2.6% vs 6.9 ± 1.6% for neutrophils, P = .0025, Figure 2A; supplemental Figure 2A). These results illustrate the proliferative advantage conferred by the JAK2V617F mutation to myeloid cells. GFP+ cells were also detectable in a small lymphoid population, with a similar level between PF4iCre;mT/mG;JAK2WT/WT and PF4iCre;mT/mG;JAK2V617F/WT mice (supplemental Figure 2B). As expected, in the bone marrow, the majority of MK expressed GFP in JAK2WT (59.2 ± 13.2%) and JAK2V617F mice (70.1 ± 6.1%). In JAK2WT mice, only a minority of erythroid (8.5 ± 1.3%) and granulocytic precursors (1.2 ± 0.2%) were GFP+, whereas in JAK2V617F mice we observed a higher frequency of GFP+ cells in erythroid (29.0 ± 8.3%, P = .05) and granulocytic (5.5 ± 1.2%) compartments (Figure 2B; supplemental Figure 2C). The low proportion of Ter119+GFP+ in the blood is probably linked to the absence of nucleus in mature erythrocytes. Indeed, the absence of new protein synthesis in erythrocytes is probably responsible for the disappearance of the protein from the membrane. This is consistent with the absence of detection of tdTomato at the surface of Ter119+ cells as previously described.14 Ter119+GFP+ cells in PF4iCre;mT/mG;JAK2V617F/WT mice probably represent the reticulocytes in which messenger RNA are still present, allowing a residual production of GFP.
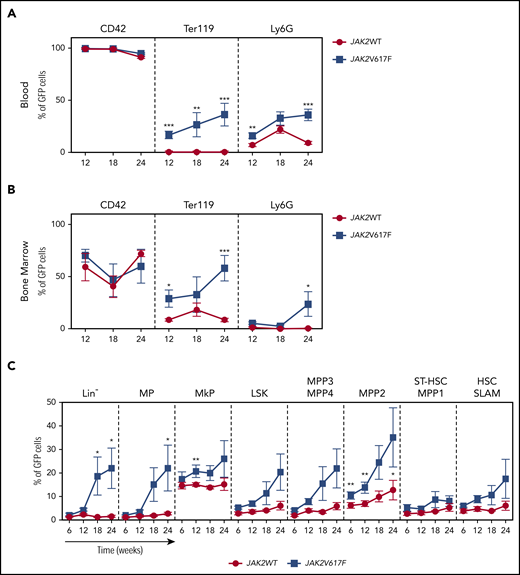
Kinetics of expansion of JAK2-mutated cells in PF4iCre;mT/mG;JAK2V617F/WTmice. Frequencies of GFP-expressing cells were assessed in the different cell populations in the blood (panel A, n = 4-12 JAK2WT; n = 5-14 JAK2V617F), in the bone marrow precursors (panel B, n = 5-9 JAK2WT; n = 4-5 JAK2V617F), and progenitors (panel C, n = 3-7 JAK2WT; n = 3-7 JAK2V617F). Data are presented as mean ± standard error of the mean. Statistical significance determined by unpaired t test. *P < .05, **P < .01, ***P < .001.
Kinetics of expansion of JAK2-mutated cells in PF4iCre;mT/mG;JAK2V617F/WTmice. Frequencies of GFP-expressing cells were assessed in the different cell populations in the blood (panel A, n = 4-12 JAK2WT; n = 5-14 JAK2V617F), in the bone marrow precursors (panel B, n = 5-9 JAK2WT; n = 4-5 JAK2V617F), and progenitors (panel C, n = 3-7 JAK2WT; n = 3-7 JAK2V617F). Data are presented as mean ± standard error of the mean. Statistical significance determined by unpaired t test. *P < .05, **P < .01, ***P < .001.
Besides, the mTmG reporter allowed us to demonstrate that the KI only occurs in the hematopoietic system but not in endothelial cells, as confirmed by the absence of detection of JAK2 mutation in sorted CD31+ cells (Figure 1D-E; supplemental Figure 1B).
The PF4 promoter induces recombination in a small subset of HSC
We then explored the recombination events at early stages of hematopoiesis (Figure 2C; supplemental Figure 2D). At 12 weeks of age, in PF4iCre;mT/mG;JAK2WT/WT mice, GFP+ cells were detectable mainly in MK progenitors (MkP, 15.1 ± 0.7%) and multipotent progenitors 2 (MPP2, 7.0 ± 1.2%, Figure 2C). In contrast, GFP+ cells were rare in other hematopoietic compartments, including short term–HSC (3.0 ± 0.7%) and SLAM-HSC (4.7 ± 1%). In PF4iCre;mT/mG;JAK2V617F/WT mice, we detected higher level of GFP+ cells in the different hematopoietic populations albeit only significant for MkP and MPP2 cells (20.8 ± 2.6%, P = .007, and 13.8 ± 2.4, P = .002, respectively) suggesting that in PF4iCre;mT/mG;JAK2V617F/WT mice, the JAK2 mutation is preferentially expressed in MPP2 and MkP, previously described as “megakaryocytes-primed populations.”19,20 Of note, we observed a nearly twofold increase in the GFP+ SLAM-HSC (8.9 ± 1.5%) compared with control mice (Figure 2C; supplemental Figure 2D). Altogether, these results demonstrate that in PF4iCre;mT/mG;JAK2V617F/WT mice, the recombination occurred not only in the megakaryocytic lineage but also in a small subset of HSC because of the leakiness of the PF4 promoter in HSC, as previously suggested.13,14 The discrepancy with recently described PF4-Cre driven JAK2V617F KI model15 ,16 could once again highlight the impact of the genetic background on the phenotypic expression of a mouse model. Moreover, the comparison of the proportion of recombined cells according to quantitative polymerase chain reaction and flow cytometry analyses points to a possible underestimation by the mTmG reporter, which could be more pronounced in the model of Woods et al compared with ours.
PF4iCre;mT/mG;JAK2V617F/WT mice allow analysis of JAK2V617F clonal dominance
Finally, we studied the kinetics of expansion of hematopoietic cells when they express JAK2V617F. In blood cells (Figure 2A) as well as in bone marrow precursors (Figure 2B), the proportion of GFP+ cells remained stable in control mice from 12 to 24 weeks in all lineages. In contrast, we observed an increase of the frequency of Ter119+GFP+ and Ly6G+GFP+ cells in mutant mice, consistent with the PV phenotype observed in these animals. A progressive amplification of nearly all categories of immature progenitors was observed in PF4iCre;mT/mG;JAK2V617F/WT mice (Figure 2C). We did not notice any significant amplification of HSC in PF4iCre;mT/mG;JAK2V617F/WT, but a high variability among mice (Figure 2C; supplemental Figure 2D). This is consistent with other mouse models and the clonal architecture observed in ET and PV patients that suggested a minor impact of the mutation on the more immature compartments.3,4
In conclusion, in PF4iCre;mT/mG;JAK2V617F/WT mice, the JAK2V617F mutation occurs in a few HSC and in a majority of MK leading to the development of a full-blown MPN. This KI model is the first to recapitulate the clonal architecture (few mutated HSC, and expanded population of mutated progenitors) observed in ET and PV (but not primary myelofibrosis) patients without using any deleterious irradiation or transplantation. Thus it represents a precious tool to study the mechanisms involved in disease initiation, clonal dominance and evolution to myelofibrosis.
For original data, please contact the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Jean Luc Villeval for the kind gift of JAK2V617F KI mice and Christian Gachet for the PF4-iCre mice.
This project was supported by research grants from ANR-DFG JAKPOT (no. ANR-14-CE35-0022-02), INSERM, and The Fondation Bettencourt Schueller, the Aquitaine Region. A.V.G. was supported by la ligue nationale contre le cancer and a research starting grant from INSERM and V.G.L. by ANR-DFG JAKPOT. I.P. was funded by INCA-PLBIO 2017.
Authorship
Contribution: C.J. designed the study and analyzed data; B.K., O.M., V.G.-L., and C.M. performed animal experimentation, flow cytometry, and analyzed data; O.M. and A.V.G. prepared the figures; O.M., A.V.G., C.J., and W.V. wrote the paper; C.J., I.P., and W.V. supervised the study; and all authors revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Chloe James, INSERM UMR 1034, CHU Haut-Lévêque, 1 Av Magellan, 33600 Pessac, France; e-mail: chloe.james@inserm.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal