

Key Points
A novel JAK2 insertion/deletion mutation is associated with eosinophilia and erythrocytosis, possibly representing a new clinical entity.
JAK2ex13InDel leads to constitutive activation and promotes signaling through β common chain–based receptors in the absence of ligand.

Abstract
The V617F mutation in the JH2 domain of Janus kinase 2 (JAK2) is an oncogenic driver in several myeloproliferative neoplasms (MPNs), including essential thrombocythemia, myelofibrosis, and polycythemia vera (PV). Other mutations in JAK2 have been identified in MPNs, most notably exon 12 mutations in PV. Here, we describe a novel recurrent mutation characterized by a common 4-amino-acid deletion and variable 1-amino-acid insertion (Leu583-Ala586DelInsSer/Gln/Pro) within the JH2 domain of JAK2. All 4 affected patients had eosinophilia, and both patients with Leu583-Ala586DelInsSer fulfilled diagnostic criteria of both PV and chronic eosinophilic leukemia (CEL). Computational and functional studies revealed that Leu583-Ala586DelInsSer (herein referred to as JAK2ex13InDel) deregulates JAK2 through a mechanism similar to JAK2V617F, activates signal transducer and activator of transcription 5 and extracellular signal-regulated kinase, and transforms parental Ba/F3 cells to growth factor independence. In contrast to JAK2V617F, JAK2ex13InDel does not require an exogenous homodimeric type 1 cytokine receptor to transform Ba/F3 cells and is capable of activating β common chain family cytokine receptor (interleukin-3 receptor [IL-3R], IL-5R, and granulocyte-macrophage colony stimulating factor receptor) signaling in the absence of ligand, with the maximum effect observed for IL-5R, consistent with the clinical phenotype of eosinophilia. Recognizing this new PV/CEL-overlap MPN has significant clinical implications, as both PV and CEL patients are at high risk for thrombosis, and concomitant cytoreduction of red cells, neutrophils, and eosinophils may be required for prevention of thromboembolic events. Targeted next-generation sequencing for genes recurrently mutated in myeloid malignancies in patients with unexplained eosinophilia may reveal additional cases of Leu583-Ala586DelInsSer/Gln/Pro, allowing for complete characterization of this unique MPN.
Introduction
JAK2V617F, a somatic mutation in the autoinhibitory JH2 (pseudokinase) domain of the non–receptor tyrosine kinase Janus kinase 2 (JAK2), promotes kinase activation and is a common driver in the classical Philadelphia chromosome–negative myeloproliferative neoplasms (MPNs), polycythemia vera (PV), essential thrombocythemia, and myelofibrosis.1,2 Rare PV cases, characterized by isolated erythrocytosis, exhibit mutations in JAK2 exon 12 in the 5′ portion of the JH2 domain.3-5 Additional non-V617F/non–exon 12 JAK2 somatic variants have been described in MPNs.6 Germline mutations other than JAK2V617F localizing to the 4.1, ezrin, radixin, moesin (FERM), JH2, or JH1 (kinase) domains of JAK2 have also been reported.7-11 JAK2 is recruited upon ligand binding to dimeric receptors, including erythropoietin (EPO) receptor (EPOR), thrombopoietin receptor (MPL), and granulocyte colony-stimulating factor receptor as well as the β common (βc) chain of granulocyte macrophage-colony stimulating factor (GM-CSF) receptor, interleukin-3 (IL-3) receptor (IL-3R), and IL-5R, resulting in kinase activation and signal transduction.12,13 In JAK2V617F, the substitution of phenylalanine for valine disrupts the autoinhibitory function of the JH2 domain by blocking JH2-mediated phosphorylation of S523 and Y570 and preventing critical conformational changes that depend on F595 and E596 in helix αC of JH2.14,15 The result is ligand-independent constitutive activation of receptors and downstream signaling via signal transduction and activation of transcription (STAT) family transcription factors. One phenotypic consequence is EPO-independent erythropoiesis, detected in vitro by formation of EPO-independent erythroid burst-forming units, known as endogenous erythroid colonies (EECs).16
Gain-of-function JAK2 mutations have been described in myeloid neoplasms other than Philadelphia chromosome–negative MPNs, including disorders with eosinophilia, most notably those characterized by a pericentriolar material 1–JAK2 rearrangement.17 JAK2V617F may occur in ≤4% of patients with hypereosinophilia of unknown significance, and survival of hypereosinophilia patients with JAK2V617F is reduced compared with those with FIP1L1-PDGFRα.12,18,19 Here, we characterize a novel insertion/deletion JAK2 mutation detected in a patient presenting with an MPN combining features of PV and chronic eosinophilic leukemia (CEL). We identified 3 additional cases of JAK2 insertion/deletion mutations involving the identical 4 residues, one of which exhibited a similar phenotype, raising the question of a specific PV/CEL-overlap syndrome associated with insertion/deletion mutations in the JAK2 JH2 domain. We demonstrate that JAK2ex13InDel bears mechanistic resemblance to JAK2V617F but can activate STAT5 in the absence of βc family cytokines IL-3, IL-5, and GM-CSF, conceivably promoting eosinophilic differentiation.
Methods
Patient samples
Written informed consent was obtained from patient 1 under The University of Utah Institutional Review Board protocol 45880. Red blood cell lysis was performed using NH4Cl/NaHCO3. Patient samples from the United Kingdom are described in supplemental Results (available at the Blood Web site).
Cell culture
The IL-3–dependent murine cell line Ba/F3 (DSMZ, Germany) was cultured in RPMI medium supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO), 2 mM l-glutamine, and 100 U/mL penicillin/streptomycin ±10% WEHI conditioned medium as a source of murine IL-3.
Construction of expression constructs and derivation of Ba/F3 lines
Standard methodology was used. See supplemental Methods for more information.
Immunoblot and immunoprecipitation
Standard methodology was used. See supplemental Methods for more information.
Measurement of drug response by cell proliferation assay
Ruxolitinib and momelotinib were purchased from Selleck Chem (Houston, TX). Ba/F3 cells expressing JAK2 mutants were seeded at 2000 cells/well in 96-well plates with graded concentrations of inhibitors in medium containing IL-3. At 72 hours, viable cells were quantified using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) reagent per the manufacturer’s instructions (Promega, Madison, WI).20 Absorbance at 490 nm was measured with an Epoch Microplate Reader (BioTek Instruments, Winooski, VT).
RNA-based analysis of X-chromosome inactivation
We used a quantitative assay based on transcript analysis of 5 X-chromosome–encoded genes informative in 95% of females.21,22 The transcription-based clonality assay was performed as previously described.23 After genotyping exonic single-nucleotide polymorphisms (SNPs) in 5 X-chromosome genes (G6PD, MPP1, FHL1, BTK, and IDS), we used quantitative allele-specific polymerase chain reaction to determine the allelic frequency of informative markers (heterozygous at the polymorphic locus being interrogated).23,24
EEC assays
We followed published protocols.24,25 See supplemental Methods for more information.
Fragment length analysis and Sanger sequencing
JAK2ex13InDel was determined using semiquantitative fragment-length analysis,21 and JAK3R925S was tested by Sanger sequencing at the DNA Sequencing Core Facility, The University of Utah. Polymerase chain reaction was performed using 20 ng DNA or complementary DNA, HotStarTaq Master Mix (Qiagen, Germantown, MD), 50 mM MgCl2, 0.2 μM of each primer (JAK2 genomic DNA: forward, 5′-FAM TTCCTACTTCGTTCTCCATCTTT-3′; reverse, 5′-TGAGAGCACATCTTTAAACAGCA-3′) (JAK2 complementary DNA: forward, 5′-FAM TGAACCAAATGGTGTTTCACA-3′; reverse, 5′-CAAATTTTACAAACTCCTGAACCA-3′) (JAK3 genomic DNA: forward, 5′-GACAGATCCTGCCTTCTCCA-3′; reverse, 5′-CAAACCACTCCTCAGCCTTC-3′).
Dual-luciferase reporter assay
See supplemental Methods for more information.
Computational modeling
The structure of JH2 with the JAK2ex13InDel mutation was predicted using the Web portal Phyre2 in intensive mode.26 The model was compared with the X-ray crystal structures of JH2 JAK2WT (Protein Data Bank [PDB]: 4FVQ) and JH2 JAK2V617F (PDB: 4FVR).27 Images were prepared in PyMOL Molecular Graphics System (DeLano Scientific, San Carlos, CA).
Statistics
Results are provided as mean ± standard error of the mean. Data were analyzed by 2-way analysis of variance with Tukey correction for multiple comparisons or a 2-tailed Student t test.
Results
Insertion/deletion mutations in the JH2 domain of JAK2 are associated with eosinophilia
A 69-year-old woman (patient 1) with a history of eosinophilic fasciitis and presumed immune thrombocytopenic purpura treated with eltrombopag presented for evaluation of steroid-refractory hypereosinophilic syndrome. She had a several-year history of peripheral blood eosinophilia with an absolute eosinophil count of ≤17 500/µL. The white blood cell count was 30 × 109/L (17.5 × 109/L eosinophils, 9.85 × 109/L neutrophils, 1.54 ×109/L lymphocytes, 1.21 × 109/L monocytes, 0.17 × 109/L immature granulocytes, and 0.14 × 109/L basophils), hemoglobin 15.7 g/dL, hematocrit (Hct) 48.3%, and platelets 193 × 109/L. Initial EPO concentration was 2.5 mU/mL (normal range, 4-27). Bone marrow biopsy specimen was hypercellular with trilineage hematopoiesis, increased atypical (hyperlobated) megakaryocytes, and markedly increased eosinophils with abnormal granulation and nuclear lobation but no increase in blasts. Cytogenetic examination showed a normal female karyotype, and the SNP microarray result was negative for copy-number alterations or copy-neutral loss of heterozygosity. The fluorescence in situ hybridization result for FIP1L1/CHIC2/PDGFRα, FGFR1, PDGFRβ, and CBFβ rearrangements was negative, as were the results of T-cell clonality studies. Next-generation sequencing (NGS) on 52 myeloid malignancy–associated genes revealed an insertion/deletion mutation in exon 13 of JAK2 (JAK2ex13InDel: Leu583-Ala586DelInsSer, c.1747_1756DelInsT) with a variant allele frequency (VAF) of 10% but no other acquired mutations (Figure 1A). Genotyping of DNA from the patient’s fingernails failed to detect JAK2ex13InDel, validating JAK2ex13InDel as a somatic mutation (NGS-identified JAK3R925S at VAF 48% confirmed to be germline). Based on the presence of 2 major criteria (Hct >48%; presence of a JAK2 mutation) and 1 minor criterion (reduced EPO), the patient fulfilled diagnostic criteria for PV while also meeting criteria for CEL.17 Computed tomography scan of the chest revealed ground glass opacities consistent with eosinophilic pulmonary involvement and a left ventricular filling defect consistent with a cardiac thrombus. The patient was placed on anticoagulation with warfarin. Ruxolitinib was started, with reduction of eosinophil counts (Figure 1B). Eltrombopag was discontinued, and repeat echocardiogram showed resolution of cardiac thrombus. Hematologic response continued for 18 months, when platelets suddenly dropped to <6 × 109/L, failed to recover upon discontinuation of ruxolitinib, and were unresponsive to a trial of steroids. Bone marrow biopsy was unchanged, without increase in blasts, and NGS continued to demonstrate JAK2ex13InDel at 9.5% VAF, with a new TET2 mutation (c.3195_3198del, pThr1066fs) at 1.6% VAF. The patient was started on 5-azacitidine, with recovery of platelet counts but persistent eosinophilia. Ruxolitinib was added, with reduction of eosinophil counts. Therapy continued with 5-azacitidine combined with ruxolitinib, with acceptable platelet and eosinophil counts.

Patient 1 JAK2 exon 13 insertion/deletion mutation and endogenous erythroid colony formation assay. (A) Structural layout of the JAK2 kinase from N terminus to C terminus. Critical domains are labeled in red. The amino acid sequences of the pseudokinase (JH2) domain of JAK2WT, JAK2V617F, and JAK2ex13InDel are highlighted. Note the deletion of residues 583 to 586 in JAK2ex13InDel and insertion of an in-frame serine residue. Tyrosine 114 in the FERM domain is critical for interactions with cytokine receptors. (B) Trends in the patient’s blood counts in response to ruxolitinib and 5-azacitidine treatment. BID, twice daily; Eos, eosinophil; Hgb, hemoglobin; Plt, platelet; WBC, white blood cell. (C) Peripheral blood mononuclear cells from the patient and a healthy control were cultured in the absence or presence of graded concentrations of EPO. In the absence of EPO, 10 EECs were observed to grow out from patient-derived mononuclear cells. Numbers above bars represent number of colonies. (D) Patient-derived EECs were plucked and genotyped using JAK2 allele-specific polymerase chain reaction followed by fragment length analysis. Seven of 8 patient-derived EECs that were genotyped exhibited heterozygosity for JAK2ex13InDel. BFU-E, erythroid burst-forming units; WT, wild-type.
Patient 1 JAK2 exon 13 insertion/deletion mutation and endogenous erythroid colony formation assay. (A) Structural layout of the JAK2 kinase from N terminus to C terminus. Critical domains are labeled in red. The amino acid sequences of the pseudokinase (JH2) domain of JAK2WT, JAK2V617F, and JAK2ex13InDel are highlighted. Note the deletion of residues 583 to 586 in JAK2ex13InDel and insertion of an in-frame serine residue. Tyrosine 114 in the FERM domain is critical for interactions with cytokine receptors. (B) Trends in the patient’s blood counts in response to ruxolitinib and 5-azacitidine treatment. BID, twice daily; Eos, eosinophil; Hgb, hemoglobin; Plt, platelet; WBC, white blood cell. (C) Peripheral blood mononuclear cells from the patient and a healthy control were cultured in the absence or presence of graded concentrations of EPO. In the absence of EPO, 10 EECs were observed to grow out from patient-derived mononuclear cells. Numbers above bars represent number of colonies. (D) Patient-derived EECs were plucked and genotyped using JAK2 allele-specific polymerase chain reaction followed by fragment length analysis. Seven of 8 patient-derived EECs that were genotyped exhibited heterozygosity for JAK2ex13InDel. BFU-E, erythroid burst-forming units; WT, wild-type.
To understand whether insertion/deletion mutations of JAK2 occur more frequently in eosinophilic conditions, we screened 173 cases from the Wessex Regional Genetics Laboratory using a custom targeted amplicon NGS covering JAK2 exon 13 (see supplemental Methods for more information on the NGS methods used). All 173 cases were negative for FIP1L1-PDGFRA, STAT5 N642H and other eosinophilia-associated abnormalities. We identified 2 additional patients with JAK2 insertion/deletion variants (VAFs 16.7% and 44%). One patient had the same mutation as the index case and presented with a dual PV/CEL clinical phenotype. A second patient had JAK2 Leu583_Ala586DelInsGln, c.1748_1756del. The identical deletion with a proline insertion was reported in a single case in an NGS study performed on patients with World Health Organization–defined hypereosinophilic syndrome and idiopathic hypereosinophilia (Table 1).28 A survey of the catalog of somatic mutations in cancer (COSMIC, accessed 28 April 2019) revealed no additional insertion/deletion mutations in this region of the JH2 domain of JAK2.
Clinical characteristics of JAK2 insertion/deletion variants
| Patient | Age (y)/sex | Variant (DNA) | Variant | VAF (%) | Additional mutations | Clinical phenotype | Clinical features |
|---|---|---|---|---|---|---|---|
| 1 | 69/F | c.1747_1756delinsT | Leu583_Ala586delinsSer | 10 | None | PV/CEL (EPO 2.5 IU/L, Hct 48.3%) | Findings: LV thrombus Treatment course: AbE improved on ruxolitinib |
| 2 | 82/F | c.1748_1757delinsA | Leu583_Ala586delinsGln | 16.70 | DNMT3A c.1728delT (VAF 32%) | CEL | Findings: cortical basal degeneration Treatment course: started on co-careldopa; deceased |
| 3 | 30/M | c.1747_1756delinsT | Leu583_Ala586delinsSer | 44 | None | PV/CEL (EPO 1.4 IU/L; Hct 57%) | Findings: visual symptoms Treatment course: Hct improved on pegylated-IFN, but no improvement in AbE |
| 4* | Unknown | c.1748_1756del | Leu583_Ala586delinsPro | N/A | N/A | CEL (unknown if PV) | Unknown |
| Patient | Age (y)/sex | Variant (DNA) | Variant | VAF (%) | Additional mutations | Clinical phenotype | Clinical features |
|---|---|---|---|---|---|---|---|
| 1 | 69/F | c.1747_1756delinsT | Leu583_Ala586delinsSer | 10 | None | PV/CEL (EPO 2.5 IU/L, Hct 48.3%) | Findings: LV thrombus Treatment course: AbE improved on ruxolitinib |
| 2 | 82/F | c.1748_1757delinsA | Leu583_Ala586delinsGln | 16.70 | DNMT3A c.1728delT (VAF 32%) | CEL | Findings: cortical basal degeneration Treatment course: started on co-careldopa; deceased |
| 3 | 30/M | c.1747_1756delinsT | Leu583_Ala586delinsSer | 44 | None | PV/CEL (EPO 1.4 IU/L; Hct 57%) | Findings: visual symptoms Treatment course: Hct improved on pegylated-IFN, but no improvement in AbE |
| 4* | Unknown | c.1748_1756del | Leu583_Ala586delinsPro | N/A | N/A | CEL (unknown if PV) | Unknown |
AbE, absolute eosinophil count; F, female; IFN, interferon; LV, left ventricular; M, male; N/A, not available.
*Pardanani et al.28
EEC formation and hypersensitivity to EPO are hallmarks of PV.16,29 Peripheral blood mononuclear cells from patient 1 were cultured in the presence of increasing concentrations of EPO.29 The erythroid progenitor cells grew EECs, and the patient’s erythroid burst-forming units were hypersensitive to EPO (Figure 1C). Genotyping of colonies revealed heterozygosity for JAK2ex13InDel in 7 of 8 EECs and JAK2WT in the remaining colony (Figure 1D). In contrast to PV, where most EECs are homozygous for JAK2V617F because of somatic uniparental disomy,30 loss of heterozygosity of JAK2ex13InDel was not detected, consistent with normal SNP array results.24,30,31
To test whether the patient’s JAK2WT cells were also clonal, we used an RNA-based clonality assay.32 Genomic DNA from the patient’s mononuclear cells was genotyped for exonic SNPs in 5 X-chromosomal genes (G6PD, MPP1, FHL1, BTK, and IDS) and found to be heterozygous for G6PD (C/T, coding sequence no. 1311, dbSNP: rs2230037) and MPP1 (G/T, coding sequence no. 358, dbSNP: rs1126762) and thus informative for a transcriptionally based clonality assay.32 Analysis of hematopoietic lineages revealed clonal platelets, neutrophils, and eosinophils (Table 2). Fragment analysis of neutrophils and eosinophils showed JAK2ex13InDel allele frequencies of 15% and 45%, respectively, indicating that JAK2ex13InDel is present in virtually all eosinophils but only a subset of neutrophils. This suggests that clonal hematopoiesis in this patient is driven by ≥1 unknown somatic mutations and that subsequent acquisition of JAK2ex13InDel biases myelopoiesis toward eosinophil differentiation. The clonal background upon which JAK2V617F mutations occur has been similarly postulated to influence MPN phenotype.24,33,34 Colony assays using mononuclear cells from our patient and healthy controls did not reveal a bias toward the formation of BFU-E (burst-forming unit-erythroid), granulocyte-macrophage colony-forming units, or granulocyte, erythrocyte, monocyte, and megakaryocyte colony-forming units (supplemental Figure 1), but results should be interpreted with caution given the variation in colony type and number observed among controls.
Summary of clonality and genotyping studies
| Cell type/source | Clonality studies with informative loci | JAK2 genotype (transcript %) | ||
|---|---|---|---|---|
| MPP1 [G/T] | G6PD [C/T] | JAK2ex13InDel | JAK2WT | |
| Granulocytes | [1/99] | [99.95/0.05] | 39 | 61 |
| Neutrophils | [3/97] | [99.75/0.25] | 15 | 85 |
| Eosinophils | [0.1/99.9] | [99.99/0.01] | 45 | 55 |
| Platelets | [4/96] | [99.5/0.5] | N/A | N/A |
| Lymphocytes | N/A | N/A | 0 | 100 |
| Cell type/source | Clonality studies with informative loci | JAK2 genotype (transcript %) | ||
|---|---|---|---|---|
| MPP1 [G/T] | G6PD [C/T] | JAK2ex13InDel | JAK2WT | |
| Granulocytes | [1/99] | [99.95/0.05] | 39 | 61 |
| Neutrophils | [3/97] | [99.75/0.25] | 15 | 85 |
| Eosinophils | [0.1/99.9] | [99.99/0.01] | 45 | 55 |
| Platelets | [4/96] | [99.5/0.5] | N/A | N/A |
| Lymphocytes | N/A | N/A | 0 | 100 |
N/A, not available.
JAK2ex13InDel confers cytokine-independent growth to Ba/F3 cells
Parental Ba/F3 cells lacking exogenous type I homodimeric cytokine receptors were transduced with human JAK2 retroviral constructs for expression of JAK2ex13InDel, JAK2V617F, or JAK2WT and cultured with IL-3 supplementation. At 48 hours after infection, green fluorescent protein (GFP)+ cells were assessed by fluorescence-activated cell sorting, and percentages were found to be comparable across genotypes in 3 independent experiments (supplemental Table 2). Following confirmation of GFP positivity, IL-3 was withdrawn and the culture monitored for GFP by flow cytometry. JAK2ex13InDel-expressing cells rapidly expanded, reaching close to 100% after 5 days (Figure 2A). In contrast, JAK2V617F-expressing cells reached only 25% GFP positivity after 10 days. In a separate set of experiments, parental Ba/F3 cells and GFP-sorted Ba/F3 cells expressing JAK2 constructs were plated at equal cell numbers and grown with or without IL-3. Parental Ba/F3 cells and those expressing JAK2WT or JAK2V617F failed to proliferate in the absence of IL-3, while JAK2ex13InDel cells demonstrated exponential growth under both medium conditions (Figure 2B). Only Ba/F3 JAK2ex13InDel cells exhibited colony growth in the absence of IL-3 (supplemental Figure 2). Parental, JAK2WT, and JAK2V617F Ba/F3 cells also displayed markedly different growth rates in response to graded concentrations of IL-3, while the growth of Ba/F3 JAK2ex13InDel was relatively insensitive to varying IL-3 dose (Figure 2C). Lastly, we performed cell proliferation experiments with ruxolitinib or momelotinib in the presence of IL-3. The 50% inhibitory concentration (IC50) of ruxolitinib and momelotinib was increased 10-fold and 2-fold, respectively, in JAK2ex13InDel Ba/F3 cells compared with controls cultured in IL-3 containing medium (Figure 2D). The inhibitory effect of ruxolitinib on growth of Ba/F3 JAK2ex13InDel cells was augmented by the absence of IL-3 (supplemental Figure 3). Altogether, these experiments demonstrate that JAK2ex13InDel potently transforms parental Ba/F3 cells to cytokine independence, with superior transforming capacity compared with JAK2V617F and resistance to JAK2 inhibitors.
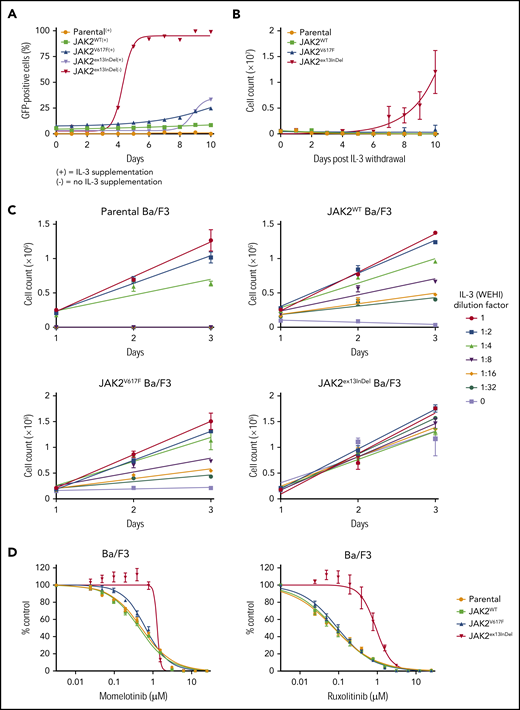
JAK2ex13InDel confers cytokine-independent growth to Ba/F3 cells. (A) Parental Ba/F3 cells were transduced with human JAK2-GFP retroviral constructs for expression of JAK2WT, JAK2V617F, or JAK2ex13InDel and cultured in WEHI conditioned medium as a source of IL-3. At 48 hours after transduction, IL-3 withdrawal led to rapid selection of JAK2ex13InDel-expressing cells (n = 3). (B) Parental Ba/F3 cells and GFP-sorted Ba/F3 cells expressing JAK2 constructs were plated at equal numbers and grown with or without IL-3. All cells proliferated in the presence of IL-3 (not shown), while only those cells containing JAK2ex13InDel exhibited exponential growth in the absence of IL-3 (n = 3). (C) Parental Ba/F3 cells and JAK2WT- and JAK2V617F-expressing Ba/F3 cells display growth sensitivity to varying concentrations of IL-3, while growth of Ba/F3 JAK2ex13InDel cells is not affected by IL-3 concentration (n = 3). (D) To assess the effect of JAK2 inhibition on Ba/F3 cells, we performed cell proliferation experiments with JAK inhibitors. The IC50 of momelotinib was increased 2-fold in JAK2ex13InDel Ba/F3 cells (n = 3). The IC50 of ruxolitinib was increased 10-fold in JAK2ex13InDel Ba/F3 cells compared with controls and JAK2V617F Ba/F3 cells.
JAK2ex13InDel confers cytokine-independent growth to Ba/F3 cells. (A) Parental Ba/F3 cells were transduced with human JAK2-GFP retroviral constructs for expression of JAK2WT, JAK2V617F, or JAK2ex13InDel and cultured in WEHI conditioned medium as a source of IL-3. At 48 hours after transduction, IL-3 withdrawal led to rapid selection of JAK2ex13InDel-expressing cells (n = 3). (B) Parental Ba/F3 cells and GFP-sorted Ba/F3 cells expressing JAK2 constructs were plated at equal numbers and grown with or without IL-3. All cells proliferated in the presence of IL-3 (not shown), while only those cells containing JAK2ex13InDel exhibited exponential growth in the absence of IL-3 (n = 3). (C) Parental Ba/F3 cells and JAK2WT- and JAK2V617F-expressing Ba/F3 cells display growth sensitivity to varying concentrations of IL-3, while growth of Ba/F3 JAK2ex13InDel cells is not affected by IL-3 concentration (n = 3). (D) To assess the effect of JAK2 inhibition on Ba/F3 cells, we performed cell proliferation experiments with JAK inhibitors. The IC50 of momelotinib was increased 2-fold in JAK2ex13InDel Ba/F3 cells (n = 3). The IC50 of ruxolitinib was increased 10-fold in JAK2ex13InDel Ba/F3 cells compared with controls and JAK2V617F Ba/F3 cells.
JAK2ex13InDel and JAK2V617F use similar mechanisms of constitutive kinase activation
JAK2ex13InDel is located in the N-lobe of the JH2 domain that faces the catalytically active JH1 domain, specifically forming the loop between the N-terminal regions of JH2 helix αC and the β3 strand. This loop is structurally close to V617F and JH2 αC26,35,36 (Figure 3A). To understand how JAK2ex13InDel leads to constitutive activation of JAK2, we modeled the effect of the L583-A586 deletion based on the structure of the JH2 domain of JAK2 (PDB: 4FVQ) using Phyre2 (intensive mode). Compared with JAK2WT, the JAK2V617F JH2 exhibits a rigid α helix C with an extra N-terminal turn. Strikingly, a similar conformational change is observed in the structural model of the JAK2ex13InDel mutant in which deletion of L583-A586 modifies the conformation of the loop between the JH2 αC and β3 strand, thereby altering the N-terminal part of αC. It has been shown previously that the autoinhibitory function of the JH2 domain can be restored in JAK2V617F via modulation of residues within the JH2 αC, specifically through mutagenesis of residue E596 to positively charged residues such as E596R/K.14 Based on our structural modeling, we predicted that JAK2ex13InDel uses the same activation mechanism as JAK2V617F. To test this, we generated Ba/F3 cells expressing JAK2E596R, JAK2E596R/V617F, and JAK2E596R/ex13InDel. In contrast to JAK2ex13InDel, JAK2E596R/ex13InDel did not exhibit IL-3–independent growth (Figure 3B). This indicates that JAK2ex13InDel belongs to the same mechanistic circuit as JAK2V617F and that mutations that eliminate or reverse the charge of E596 disrupt both JAK2V617F and JAK2ex13InDel-mediated constitutive kinase activation. It is conceivable that additional mechanisms enhance constitutive kinase activation. For instance, molecular dynamics simulation suggested that phosphorylation of Y570 participates in the maintenance of inhibitory interactions between JH1 and JH2.35 In JAK2ex13InDel, the altered position of Y570 may disrupt inhibitory interactions to a greater degree than in JAK2V617F. Increased kinase activity could explain the reduced sensitivity of JAK2ex13InDel to JAK2 inhibitors. Unfortunately, repeated attempts to directly compare enzyme kinetics between full-length human JAK2WT, JAK2ex13InDel, and JAK2V617F were unsuccessful, as we were unable to achieve sufficient concentrations of full-length purified protein, an experimental hurdle known in the field.37
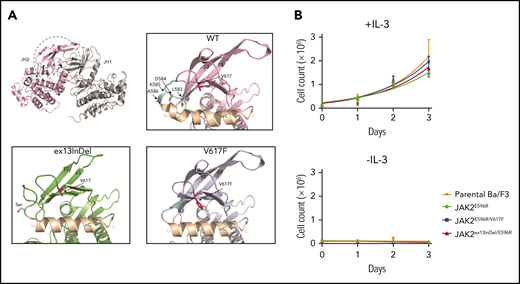
JAK2ex13InDel and JAK2V617F use similar mechanisms of constitutive kinase activation. (A) Residues L583-A586 are located in the N-lobe of the pseudokinase domain of JAK2 (JH2) that faces the catalytically active kinase domain (JH1) (top left). L583-A586 forms the loop between the N-terminal JH2 αC and the β3 strand in JAK2WT (top right, light blue). This loop is structurally close to V617 and the JH2 αC, which is altered in the presence of the V617F mutation. Compared with the loose conformation of JH2 αC in JAK2WT (bottom right), the JH2 αC of JAK2V617F exhibits a rigid helix (bottom right). A strikingly similar conformational change in JH2 αC is observed in the JAK2ex13InDel mutant (bottom left). (B) To demonstrate that E596 is also critical for JAK2ex13InDel activity, we cultured Ba/F3 cells expressing JAK2E596R, JAK2E596R/V617F, and JAK2ex13InDel/E596R with or without IL-3. JAK2ex13InDel/E596R did not demonstrate IL-3–independent growth (n = 3).
JAK2ex13InDel and JAK2V617F use similar mechanisms of constitutive kinase activation. (A) Residues L583-A586 are located in the N-lobe of the pseudokinase domain of JAK2 (JH2) that faces the catalytically active kinase domain (JH1) (top left). L583-A586 forms the loop between the N-terminal JH2 αC and the β3 strand in JAK2WT (top right, light blue). This loop is structurally close to V617 and the JH2 αC, which is altered in the presence of the V617F mutation. Compared with the loose conformation of JH2 αC in JAK2WT (bottom right), the JH2 αC of JAK2V617F exhibits a rigid helix (bottom right). A strikingly similar conformational change in JH2 αC is observed in the JAK2ex13InDel mutant (bottom left). (B) To demonstrate that E596 is also critical for JAK2ex13InDel activity, we cultured Ba/F3 cells expressing JAK2E596R, JAK2E596R/V617F, and JAK2ex13InDel/E596R with or without IL-3. JAK2ex13InDel/E596R did not demonstrate IL-3–independent growth (n = 3).
JAK2ex13InDel, but not JAK2V617F, activates ERK1/2 and STAT5 signaling in Ba/F3 cells in the absence of IL-3
We next assessed canonical signaling pathways activated by JAK2V617F.38 Ba/F3 cells expressing JAK2ex13InDel, JAK2V617F, or JAK2WT maintained in IL-3 were subjected to an extensive washout protocol to remove IL-3 and then replated with or without IL-3, followed by immunoblot analysis of whole-cell extracts. As expected, all cell lines cultured in the presence of IL-3 demonstrated robust activation of extracellular signal-regulated kinase 1/2 (ERK1/2), STAT5, and JAK2 (Figure 4A-B). In contrast, activation of ERK1/2, STAT5, and JAK2 persisted in Ba/F3 cells expressing JAK2ex13InDel despite IL-3 withdrawal. No consistent differences were observed for STAT3, SHP2, or p38 mitogen-activated protein kinase (Figure 4). JAK2 tyrosine phosphorylation was readily demonstrable in JAK2 immunoprecipitates from all cell lines when cultured with IL-3. In the absence of IL-3, only JAK2ex13InDel showed weak but reproducible tyrosine phosphorylation. As activation of downstream signaling by JAK2V617F depends on association with a cytokine receptor, such as EPOR, the data are consistent with the lack of EPOR expression in Ba/F3 cells and suggest that JAK2ex13InDel is either capable of a receptor interaction that is not accessible to JAK2V617F or does not require such interaction.
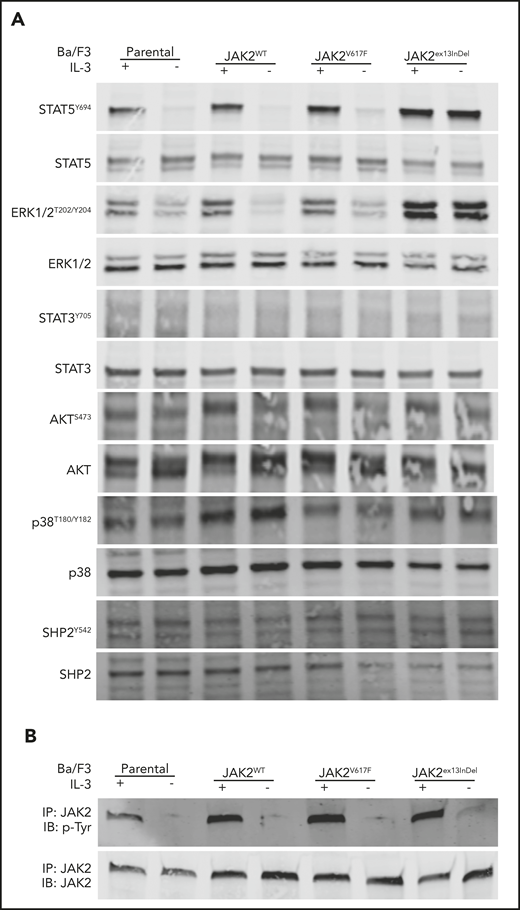
JAK2ex13InDel activates ERK1/2 and STAT5 signaling in Ba/F3 cells in the absence of IL-3. (A) Ba/F3 cells cultured in the presence of IL-3 displayed activation of STAT5 and ERK1/2. In the absence of IL-3, STAT5 and ERK1/2 activation was maintained only by JAK2ex13InDel-expressing cells. Additional proteins did not demonstrate differential JAK2ex13InDel activation in the absence of IL-3. (B) In the absence of IL-3, JAK2 phosphorylation was detectable only in JAK2ex13InDel-expressing cells. IB, immunoblot; IP, immunoprecipitate.
JAK2ex13InDel activates ERK1/2 and STAT5 signaling in Ba/F3 cells in the absence of IL-3. (A) Ba/F3 cells cultured in the presence of IL-3 displayed activation of STAT5 and ERK1/2. In the absence of IL-3, STAT5 and ERK1/2 activation was maintained only by JAK2ex13InDel-expressing cells. Additional proteins did not demonstrate differential JAK2ex13InDel activation in the absence of IL-3. (B) In the absence of IL-3, JAK2 phosphorylation was detectable only in JAK2ex13InDel-expressing cells. IB, immunoblot; IP, immunoprecipitate.
JAK2ex13InDel, but not JAK2V617F, activates signaling through βc-associated cytokine receptors
All 4 patients in our series showed eosinophilia, raising the question whether JAK2ex13InDel causes eosinophil lineage bias. As the βc family cytokines IL-3, IL-5, and GM-CSF have been shown to promote eosinophilic differentiation, we transfected HEK293 cells with the βc chain and the respective α chains of the IL-3, IL-5, and GM-CSF receptors, luciferase-based STAT5 reporters, and JAK2WT, JAK2V617F, JAK2ex13InDel, or empty vector.39 We chose HEK293 cells, as they are human derived and do not express endogenous βc receptors, allowing us to isolate signaling events.40 We assessed STAT5 transcriptional activity in the presence and absence of IL-3, IL-5, or GM-CSF (Figure 5A-C). All 3 cytokines activated STAT5 in empty vector, JAK2WT-, JAK2V617F-, and JAK2ex13InDel-expressing cells, while only JAK2ex13InDel-expressing cells displayed STAT5 activation in the absence of IL-3, IL-5, or GM-CSF, with the strongest effect observed in cells expressing IL-5R. Interestingly, the βc chain alone activated the reporter in JAK2ex13InDel expressing cells, while IL-5R α-chain alone did not, with the strongest effect observed when both were coexpressed (supplemental Figure 4). Thus, unlike JAK2WT and JAK2V617F, JAK2ex13InDel is uniquely capable of activating βc family cytokine signaling in HEK293 cells. Additional luciferase-based STAT5 assays in HEK293 cells demonstrate that both JAK2V617F and JAK2ex13InDel are capable of activating EPOR and MPL in the absence of cytokine. While EPOR activation is comparable, cytokine-independent activation of MPL by JAK2V617F is enhanced compared with JAK2ex13InDel (supplemental Figure 5).
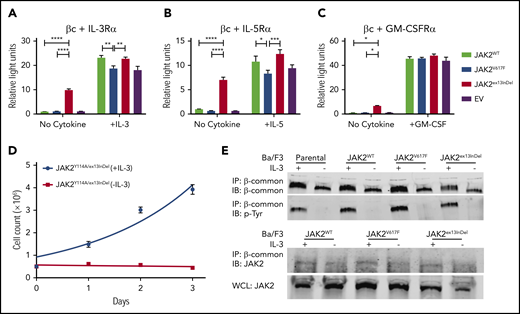
JAK2ex13InDel activates signaling through βc-associated cytokine receptors. (A-C) HEK293 cells expressing βc were transfected with the α chains of the IL-3R, IL-5R, or CSF2R (GM-CSF receptor), together with a luciferase-based STAT5 reporter (Spi_Luc) and JAK2WT, JAK2V617F, or JAK2ex13InDel. STAT5 transcriptional activity was measured in the presence or absence of the respective cytokines at 10 ng/mL (n = 3). Data were analyzed with a 2-way analysis of variance and Tukey’s correction for multiple comparisons (*P < .05, **P < .01, ***P < .001, ****P < .0001). (D) The FERM domain mutation Y114A was introduced into JAK2ex13InDel. Ba/F3 cells expressing JAK2Y114A/ex13InDel cells were cultured with or without IL-3. JAK2 double-mutant cells did not proliferate upon IL-3 withdrawal (n = 3). (E) Parental and Ba/F3 cells expressing JAK2WT, JAK2V617F, and JAK2ex13InDel grown with IL-3 supplementation were washed to remove IL-3 and replated with or without IL-3. Cells were harvested after 4 hours. βc immunoprecipitates were resolved on SDS-PAGE and incubated with a phosphotyrosine antibody (top). A representative experiment out of 3 independent repeats is shown. βc immunoprecipitates were incubated with a JAK2 antibody (bottom). A representative experiment out of 3 independent repeats is shown. WCL, whole-cell lysate.
JAK2ex13InDel activates signaling through βc-associated cytokine receptors. (A-C) HEK293 cells expressing βc were transfected with the α chains of the IL-3R, IL-5R, or CSF2R (GM-CSF receptor), together with a luciferase-based STAT5 reporter (Spi_Luc) and JAK2WT, JAK2V617F, or JAK2ex13InDel. STAT5 transcriptional activity was measured in the presence or absence of the respective cytokines at 10 ng/mL (n = 3). Data were analyzed with a 2-way analysis of variance and Tukey’s correction for multiple comparisons (*P < .05, **P < .01, ***P < .001, ****P < .0001). (D) The FERM domain mutation Y114A was introduced into JAK2ex13InDel. Ba/F3 cells expressing JAK2Y114A/ex13InDel cells were cultured with or without IL-3. JAK2 double-mutant cells did not proliferate upon IL-3 withdrawal (n = 3). (E) Parental and Ba/F3 cells expressing JAK2WT, JAK2V617F, and JAK2ex13InDel grown with IL-3 supplementation were washed to remove IL-3 and replated with or without IL-3. Cells were harvested after 4 hours. βc immunoprecipitates were resolved on SDS-PAGE and incubated with a phosphotyrosine antibody (top). A representative experiment out of 3 independent repeats is shown. βc immunoprecipitates were incubated with a JAK2 antibody (bottom). A representative experiment out of 3 independent repeats is shown. WCL, whole-cell lysate.
The ability of JAK2V617F to transform hematopoietic cells requires interaction with a cytokine receptor,12 while certain JAK3 mutants have been shown to activate signal transduction without the need for interaction with a cytokine receptor.41 As tyrosine 114 in the N-terminal FERM domain of JAK2 is essential for cytokine receptor interactions,41-43 we introduced the Y114A mutation into JAK2ex13InDel and expressed the double mutant in Ba/F3 cells. Sorted Ba/F3 JAK2Y114A/ex13InDel cells were cultured with or without IL-3 for 3 days. Ba/F3 cells expressing JAK2Y114A/ex13InDel did not proliferate in the absence of IL-3, while exponential growth was observed with IL-3, suggesting that cellular transformation and oncogenic signaling by JAK2ex13InDel is dependent upon association with a cytokine receptor (Figure 5D). While JAK2V617F typically associates with a homodimeric type I cytokine receptor, components of certain heterodimeric receptors, including the βc of the GM-CSF receptor, IL-3R, and IL-5R, can cooperate with JAK2V617F to activate downstream signaling and induce cellular transformation.44 Given the differential ability of JAK2ex13InDel compared with JAK2V617F to activate heterodimeric receptor signaling in HEK293 cells (Figure 5A-C), we hypothesized that JAK2ex13InDel may differentially bind to and phosphorylate βc in Ba/F3 cells. To test this, we assessed tyrosine phosphorylation of βc immunoprecipitates from Ba/F3 cells expressing JAK2WT, JAK2V617F, or JAK2ex13InDel. We consistently detected JAK2 in βc immunoprecipitates from cells growing in IL-3, while no interaction was identified in the absence of IL-3, irrespective of genotype (Figure 5E, bottom panel; supplemental Figure 6). In accord with this, we saw no consistent βc phosphorylation in the absence of IL-3, and phosphorylation in the presence of IL-3 was comparable across genotypes (Figure 5E, top panel). These data suggest that transformation of Ba/F3 cells by JAK2ex13InDel may use another as-yet-unidentified cytokine receptor.
Discussion
We describe for the first time the structure and function of a novel JAK2 exon 13 insertion/deletion mutant identified in a patient fulfilling diagnostic criteria for both PV and CEL. Consistent with the clinical PV phenotype, mononuclear cells from the patient demonstrate EPO hypersensitivity and EEC formation, phenocopying JAK2V617F. However, unlike in PV subjects with JAK2V617F mutation wherein somatic uniparental disomy is detected in most EEC clones,24,30 we detected not homozygosity of JAK2ex13InDel in our patient’s EECs but heterozygosity. This finding may account for the relatively low proportion of EECs in our patient compared with those observed in classic PV. Further, JAK2ex13InDel imparts IL-3–independent growth to Ba/F3 cells in the absence of an exogenous cytokine receptor, resulting in constitutive STAT5 and ERK1/2 activation. Compared with JAK2V617F, the transforming potency of JAK2ex13InDel toward Ba/F3 cells is significantly increased in multiple assays (Figure 2A-D). There are 2 possible explanations for this striking difference. First, JAK2ex13InDel may have increased intrinsic kinase activity compared with JAK2V617F due to conformational differences. While JAK2ex13InDel operates within the same activation circuit as JAK2V617F, our modeling suggests that unique conformational attributes may mitigate residual autoinhibitory Y570 interactions present in JAK2V617F, thereby potentiating JAK2ex13InDel transformation capacity. The reduced sensitivity of JAK2ex13InDel to JAK kinase inhibitors would also be consistent in principle with this notion (Figure 2D) but could also be explained by differences in inhibitor binding affinity to the catalytic site. Unfortunately, we were unable to directly compare kinase activity across genotypes, as multiple attempts to generate sufficient quantities of recombinant full-length JAK2 proteins were unsuccessful.
An alternative explanation is that JAK2ex13InDel may be able to interact with a cytokine receptor or other signaling molecule inaccessible to JAK2V617F. Ba/F3 cells expressing human JAK2ex13InDel were transformed to cytokine independence in the absence of an exogenous receptor, while Ba/F3 cells expressing JAK2V617F were not, consistent with previous reports that coexpression of a homodimeric cytokine receptor is required for JAK2V617-mediated Ba/F3 transformation.12 This is surprising in view of our computational modeling that supports analogous mechanisms of kinase activation in JAK2ex13InDel and JAK2V617F (Figure 3A) and suggests that JAK2ex13InDel has acquired additional functional capabilities. In accord with this idea, JAK2ex13InDel increases STAT5 transcriptional activity in HEK293 cells expressing IL-3R, IL5-R, or GM-CSFR, while JAK2WT and JAK2V617F have no effect (Figure 5A-C). This indicates that JAK2ex13InDel can activate the βc family of receptors to induce cytokine-independent STAT5 signaling. Remarkably, effects were most pronounced for IL-5R, the cytokine with the strongest association with eosinophil differentiation, providing a potential mechanistic link to the eosinophilia observed in patients with JAK2 Leu583-Ala586DelInsSer/Gln/Pro. Altogether, our data suggest that alterations within the JH2 domain can affect JAK2 functionalities, in addition to autoinhibition of kinase activity, and that JAK2ex13InDel may skew differentiation toward the eosinophil lineage through ligand-independent activation of IL-5/STAT5 signaling. As JAK2ex13InDel also activates IL-3 and GM-CSF signaling, and synergism among the 3 βc cytokines is crucial to optimal eosinophilic growth and differentiation, JAK2ex13InDel may be uniquely capable of inducing eosinophil differentiation and growth without the need for a cooperating mutation.39,45-48 Consistent with this, our patient’s JAK2ex13InDel allele frequency was highest in eosinophils, which indicates that the mutation per se enhances eosinophil expansion following the establishment of clonal hematopoiesis by an as-yet-undetermined pre-JAK2 mutational event (Figure 1D). While it is possible that an antecedent genetic event promotes the eosinophilic hematopoiesis observed with JAK2ex13InDel, our functional studies suggest that the signaling properties of JAK2ex13InDel drive hypereosinophilia. Several attempts to test this in primary CD34+ cells were unsuccessful, as we were unable to trace eosinophilic differentiation due to consistent loss of reporter signal from transduced cells (supplemental Figure 7). Further investigation will be required to characterize the JAK2ex13InDel interactome and its unique features compared with JAK2V617F to precisely delineate the relationship between JAK2ex13InDel and eosinophilic lineage bias.
Our patient’s hypereosinophilia partially responded to ruxolitinib; however, dose intensity was limited by thrombocytopenia (Figure 1C). Although thrombocytopenia is a well-known side effect of ruxolitinib,49 in our patient, it may also reflect a feature of this clonal myeloid disorder. This is supported by abnormal megakaryocyte morphology and the fact that platelet counts improved on 5-azacitidine. As such, the preceding diagnosis of immune thrombocytopenic purpura may have been incorrect. We identified 3 additional cases of JAK2 insertion/deletion mutations within the JH2 domain, 1 identical to patient 1 (Leu583-Ala586DelInsSer) and 2 with deletion of the same 4 amino acids, but with insertion of proline or glutamine rather than serine (Leu583_Ala586DelInsPro/Gln). All 4 patients had eosinophilia. In contrast, JAK2V617F is rare in patients with idiopathic hypereosinophilia, accounting for only 4% of cases in large series.18 The fact that, to the best of our knowledge, no additional insertion/deletion mutation in this region of the JAK2 JH2 domain has been described strongly suggests a specific genotype-phenotype correlation, in contrast to the phenotypic promiscuity of JAK2V617F. Moreover, 2 of 3 cases with available data, including both with Leu583-Ala586DelInsSer, were associated with a polycythemia and low EPO, suggesting that JAK2ex13InDel may cause a previously unrecognized clinical syndrome that combines features of PV and CEL. Like JAK2V617F, JAK2ex13InDel promotes EEC formation (Figure 1D), yet only JAK2ex13InDel is capable of co-opting βc family cytokine receptor signaling to induce cytokine-independent STAT5 activity in HEK293 cells (Figure 5A). Due to concomitant polycythemia and eosinophilia, JAK2ex13InDel patients may have an especially high risk of thrombosis, warranting screening with echocardiogram and/or computed tomography scans to detect occult vascular complications, such as the cardiac thrombus in our patient. Although this is the first characterization of a JAK2ex13InDel somatic mutation, the routine use of NGS in MPN patients may reveal additional cases and shed further light on the spectrum of clinical phenotypes associated with JAK2 insertion/deletion mutations. As insertion/deletion mutations are more difficult to detect by NGS and the JAK2 mutations described here are subclonal, similar to most JAK2V617F PVs, this report underlines the need careful inspection of sequencing traces.
For original data, please contact the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors acknowledge the research contributions of patient 1 and thank the clinical staff dedicated to her care, particularly Karissa Dimas and Jeffrey Gilreath.
This work was supported by National Institutes of Health, National Cancer Institute grant R01CA178397 (M.W.D. and T.O.), an LLS Specialized Center of Research Program Award (GCNCR0314A-UTAH) (M.W.D.), a V Foundation for Cancer Research Translational Research Award (M.W.D. and T.O.), the Ludwig Institute for Cancer Research (S.N.C.), Fondation contre le Cancer (S.N.C.), Salus Sanguinis (S.N.C.), and Fondation “Les avions de Sébastien” (projects Action de recherche concertée 16/21-073 and WelBio F 44/8/5 MCF/UIG 10955) (S.N.C.). The University of Utah Flow Cytometry Facility is supported by the National Institutes of Health through National Cancer Institute award 5P30CA042014-24 and the National Center for Research Resources award 1S10RR026802-01. A.B.P. is supported by an American Society of Hematology Research Training Award for Fellows. D.Y. is supported by a Special Fellow Award from the Leukemia & Lymphoma Society. The National Institutes of Health, Cancer Center Support Grant (P30 CA042014) awarded to the Huntsman Cancer Institute provided developmental funds and shared resources critical to this project.
Authorship
Contribution: A.B.P., A.F., S.N.C., T.O., J.T.P., and M.W.D. contributed to conception and design; A.B.P., A.F., E.L., S.J.K., and M.S. developed methodology; A.B.P., A.F., E.L., S.J.K., L.G., M.X., J.M.A., D.Y., and A.D.P. acquired data; A.B.P., A.F., E.L., A.D.P., S.J.K., J.T.P., M.S., N.C.P.C., S.N.C., and M.W.D. analyzed and interpreted data; A.M.A., A.B.P., J.A., J.L., M.W.D. provided patient data and material; A.B.P., A.F., E.L., S.N.C., N.C.P.C., S.J.K., G.J.G., T.O., J.T.P., and M.W.D. wrote, reviewed, and/or revised the manuscript; and P.C. provided administrative, technical, or material support.
Conflict-of-interest disclosure: M.W.D. reports research funding from and is a paid advisory board member of and/or consultant for Blueprint, Pfizer, Takeda, Ascentage Pharma, TRM, and Humana. N.C.P.C. reports research support from Novartis and is a paid advisory board member of Novartis and Incyte. S.N.C. is the co-founder of MyeloPro Research and Diagnostics, GmbH, Vienna, Austria. J.T.P. is a paid advisory board member of Agios Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Michael W. Deininger, Division of Hematology and Hematologic Malignancies, Huntsman Cancer Institute, The University of Utah, 2000 Circle of Hope Drive, Salt Lake City, UT, 84112; e-mail: michael.deininger@hci.utah.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal