

Key Points
HA catabolism is dysregulated in the endothelium of patients with IBD.
Platelet HYAL2 regulates inflammation by limiting leukocyte trans-endothelial migration.

Abstract
Platelets are specialized cells essential for hemostasis that also function as crucial effectors capable of mediating inflammatory and immune responses. These sentinels continually survey their environment and discriminate between homeostatic and danger signals such as modified components of the extracellular matrix. The glycosaminoglycan hyaluronan (HA) is a major extracellular matrix component that coats the vascular lumen and, under normal conditions, restricts access of inflammatory cells. In response to tissue damage, the endothelial HA matrix enhances leukocyte recruitment and regulates the early stages of the inflammatory response. We have shown that platelets can degrade HA from the surface of activated endothelial cells via the enzyme hyaluronidase-2 (HYAL2) and that HYAL2 is deficient in platelets isolated from patients with inflammatory bowel disease (IBD). Platelets are known to be involved in the pathogenesis of several chronic disease states, including IBD, but they have been largely overlooked in the context of intestinal inflammation. We therefore wanted to define the mechanism by which platelet HYAL2 regulates the inflammatory response during colitis. In this study, we provide evidence that HA catabolism is disrupted in human intestinal microvascular endothelial cells isolated from patients with IBD. Furthermore, mice deficient in HYAL2 are more susceptible to an acute model of colitis, and this increased susceptibility is abrogated by transfusion of HYAL2-competent platelets. Finally, we show that platelets, via HYAL2-dependent degradation of endothelial HA, regulate the early stages of inflammation in colitis by limiting leukocyte extravasation.
Introduction
Inflammatory bowel disease (IBD) is a chronic relapsing disorder comprising 2 main subtypes, ulcerative colitis (UC) and Crohn’s disease (CD), with different clinical manifestations but both ultimately leading to relentless destruction of the gastrointestinal tract. The pathogenesis of IBD is multifactorial, in which interactions between genetic and environmental factors promote immunoregulatory dysfunction and result in chronic inflammation.1,2 Evidence from animal models and human studies show that IBD involves not only dysregulated immune cells but is also driven by complex interactions between cell types, including the intestinal microvasculature and platelets.3,4
A growing body of evidence shows that platelets play direct roles in the immune response and inflammatory cascade.5 Platelet abnormalities are a well-established clinical feature of IBD, in which the blood of patients exists in a hypercoagulable state.6,7 Increased platelet number and reactivity associate with serologic disease markers in IBD, and platelets are known to circulate in an activated state.8-11 Several platelet surface glycoprotein receptors are altered in IBD, and our laboratory found that the extracellular matrix (ECM)-degrading enzyme hyaluronidase-2 (HYAL2) is deficient in platelets isolated from patients with IBD.9,12,13
Tissue damage and bleeding are consistent features of IBD progression, and remodeling of ECM molecules are strongly implicated in IBD pathogenesis.14-16 The glycosaminoglycan hyaluronan (HA) is widely distributed in the ECM of many tissues and plays key roles in inflammation.17 HYAL2 belongs to a family of hyaluronidases18 but is unique in that it is the only member expressed in platelets and megakaryocytes, where it plays a role in proplatelet formation.18-20 HYAL2, a glycophosphatidylinositol-anchored protein, is packaged within a subset of platelet α granules and, upon platelet activation, is translocated to the platelet surface where it functions with coreceptors to digest HA.12,21
A dynamic glycocalyx comprising HA and other membrane-bound glycoconjugates lines the endothelial surface and restricts immune cell access under normal conditions.22 Inflammatory stimuli enhance HA synthesis and promote the formation of a form of HA that is covalently modified with the heavy chains (HCs) from the serum proteoglycan inter-α-trypsin inhibitor (IαI).23 Importantly, HA produced on the luminal surface of inflamed microvascular endothelial cells plays a key role in the adhesion and extravasation of immune cells in multiple sclerosis, liver disease, and IBD.24-29
The current article defines a novel mechanism whereby platelets are capable of limiting leukocyte extravasation during the early stages of colitis by degrading HA on the endothelial cell surface. We show that mice deficient in the enzyme HYAL2 are more susceptible to dextran sodium sulfate (DSS)-induced colitis and that this increased susceptibility can be reversed by restoring HYAL2 activity in platelets. Furthermore, we show that platelets incubated with inflamed human microvascular endothelial cells (HIMECs) are capable of reducing leukocyte extravasation via an HYAL2-dependent mechanism and that this pathway is disrupted in platelets isolated from patients with IBD.
Methods
Human tissue, cell isolation, and culture
Human intestinal tissue was obtained from the Department of Surgical Pathology at the Cleveland Clinic (Cleveland, OH). All specimens were of colonic origin, and diagnoses were confirmed by using clinical criteria. Tissues were obtained from histologically normal, noninflamed large-bowel specimens (non-IBD controls) from patients admitted for bowel resection because of conditions that included colon cancer, benign polyps, diverticulitis, and colonic inertia. HIMECs were isolated and cultured as previously reported.30,31 The study was approved by the Institutional Review Board of the Cleveland Clinic.
DSS-induced colitis
Colitis was induced in wild-type (WT) and HYAL2 null mice by supplying sterile acidified water with or without the addition of 2.5% DSS (MP Biomedicals, Solon, OH) ad libitum in water bottles. All mice were weighed and monitored daily for signs of colitis. Animals were euthanized on days 0, 4, or 7. Serum was collected by cardiac puncture. The large intestine from the cecum to the rectum was dissected and measured for length; 1 cm regions were taken from the terminal ileum, proximal, transverse, and distal colon and fixed in 10 times tissue volume of molecular biology grade Histochoice (MilliporeSigma, St. Louis, MO).
Platelet transfusion
Mouse blood was collected from the retro-orbital plexus into anticoagulant citrate dextrose solution. Platelet-rich plasma was obtained by centrifugation at 100g for 8 minutes followed by centrifugation of platelet-rich plasma at 700g in the presence of prostacyclin (2 μg/mL) for 5 minutes at room temperature. After washing once, platelets were resuspended in modified Tyrode’s buffer (137 mM NaCl, 0.3 mM Na2HPO4, 2 mM KCl, 12 mM NaHCO3, 5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 5 mM glucose, pH 7.3). Platelets from WT or HYAL2 knock-out (KO) donor mice were pooled, and the platelet count was adjusted to 4 × 109 platelets/mL. Recipient mice were injected with 200 μL via the retro-orbital plexus.
Platelet coculture trans-endothelial migration assay
The upper surface of 5.0 μm pore size Transwell filters were coated with fibronectin (Thermo Fisher Scientific, Waltham, MA) for 1 hour, rinsed with phosphate-buffered saline, and seeded with HIMECs to confluency in EGM-2 medium (Lonza, Basel, Switzerland) and cultured for 5 days at 37°C to allow for tight junction formation. HIMECs were pretreated with or without tumor necrosis factor‐α (TNF‐α) (10 ng/mL, 16 hours) with or without platelets (1 × 107 platelets, 1 hour) with or without Streptomyces hyaluronidase (1 mU/mL, 30 minutes) and washed to remove treatments. In some cases, platelets were pretreated with anti-HYAL2 antibody (25 μg/mL; Abcam, Cambridge, United Kingdom) or control immunoglobulin G (25 μg/mL). Freshly isolated human peripheral blood mononuclear cells (PBMCs) from healthy donors were labeled with Calcein AM 2 μM (1 hour on ice; Thermo Fisher Scientific) in phosphate-buffered saline containing 1% fetal bovine serum, washed, resuspended in RPMI 1640 containing 1% fetal bovine serum, and added (1 × 106 PBMCs per well) to HIMECs. Chemokine (C-C motif) ligand 5 (CCL5) was added to the lower chamber as a chemoattractant. Transmigration was allowed to proceed for 3 hours at 37°C. Calcein AM–labeled PBMCs in the lower wells after 3 hours were analyzed by using an automated Leica fluorescence microscope at 20× magnification in well-scan mode (Leica Biosystems, Buffalo Grove, IL), and the number of transmigrated PBMCs in each well was determined by using Image-Pro Plus software (Media Cybernetics, Rockville, MD).
Mice, microscopy, immunoblots, and HA analysis
Detailed protocols for mouse procedures, microscopy, immunoblots, and HA analysis are provided in the supplemental Data (available on the Blood Web site).
Results
HYAL2 KO mice are highly susceptible to DSS-induced colitis
Our previous studies showed that HA accumulates within inflamed intestinal tissue of patients with IBD and mice subjected to experimental models of colitis.19,27,32 HA deposition is an early event that precedes inflammation in the DSS-induced colitis model,26,27 and we hypothesized that defects in HA turnover might accelerate disease progression. Mice supplied with DSS in drinking water reproducibly develop colitis in a time- and concentration-dependent manner.33 DSS treatment compromises the epithelial barrier, leading to a bacterially induced colitis, and pathology is first observed in the distal colon where bacterial burden is greatest. We initiated intestinal inflammation by delivering 2.5% DSS in the drinking water of HYAL2-deficient and control mice, and disease was measured over 7 days. Loss of HYAL2 led to accelerated and increased disease, measured according to overall weight loss (Figure 1A-B; supplemental Figure 1A). Mice were evaluated for outward signs of disease activity by using the disease activity index (DAI); these signs included rectal bleeding, loose stools, ruffled fur, hunched posture, and rectal prolapse. Our data show that HYAL2 null mice were more severely affected (Figure 1C) compared with control mice (n = 10 each; P < .01). Histologic examination of distal colon sections of unchallenged mice revealed a thicker external muscle layer but no morphologic differences in the colonic mucosa in HYAL2 null mice compared with control mice (supplemental Figure 2A-B). However, after 7 days of DSS treatment, increased epithelial erosion, crypt loss, angiogenesis, submucosal swelling, and increased infiltration of immune cells were observed in HYAL2-deficient mice compared with WT control mice (Figure 1A,D; supplemental Figure 3). Changes in colon length due to DSS-induced colitis are a common indicator of disease severity in this model, and HYAL2 KO mice also exhibited a significantly shortened colon compared with control mice (Figure 1E).

HYAL2 KO mice are more susceptible to DSS-induced colitis. (A) Cross-sections of distal colon collected from mice after 7 days of colitis induction with 2.5% DSS. Hematoxylin-eosin staining highlights the cellular and structural changes in the intestine, including inflammatory leukocyte infiltration. Boxed regions are enlarged in adjacent panels. Scale bars indicate 500 μm. L denotes the intestinal lumen, triangles indicate the epithelial layer, S denotes the submucosa, and arrows indicate the muscularis mucosae. (B) Body weight was measured on day 7 and compared with starting weight. (C) Mice were scored for outward signs of disease, including weight loss, hunched posture, ruffled fur, bloody stools, and rectal prolapse. (D) Distal colon sections from mice after 7 days of DSS were scored for pathologic changes, including erosion of the epithelial layer, leukocyte infiltration, submucosal swelling, muscularis mucosae hyperplasia, and increased vascularization. (E) Colons from mice euthanized after 7 days of DSS were measured for distance (centimeters) from the distal end of the cecum to the rectum. Symbols represent individual mice. Symbols indicate genotypes: (▲) WT mice and (●) HYAL2 KO mice. Data are reported as mean ± SEM; n = 10 mice per group. *P < .05.
HYAL2 KO mice are more susceptible to DSS-induced colitis. (A) Cross-sections of distal colon collected from mice after 7 days of colitis induction with 2.5% DSS. Hematoxylin-eosin staining highlights the cellular and structural changes in the intestine, including inflammatory leukocyte infiltration. Boxed regions are enlarged in adjacent panels. Scale bars indicate 500 μm. L denotes the intestinal lumen, triangles indicate the epithelial layer, S denotes the submucosa, and arrows indicate the muscularis mucosae. (B) Body weight was measured on day 7 and compared with starting weight. (C) Mice were scored for outward signs of disease, including weight loss, hunched posture, ruffled fur, bloody stools, and rectal prolapse. (D) Distal colon sections from mice after 7 days of DSS were scored for pathologic changes, including erosion of the epithelial layer, leukocyte infiltration, submucosal swelling, muscularis mucosae hyperplasia, and increased vascularization. (E) Colons from mice euthanized after 7 days of DSS were measured for distance (centimeters) from the distal end of the cecum to the rectum. Symbols represent individual mice. Symbols indicate genotypes: (▲) WT mice and (●) HYAL2 KO mice. Data are reported as mean ± SEM; n = 10 mice per group. *P < .05.
Platelets isolated from mice with DSS-induced colitis have reduced HYAL2 protein and hyaluronidase activity
IBD is intimately associated with platelet abnormalities,34 and we hypothesized that platelets isolated from DSS-treated mice might also exhibit reduced HA-degrading activities. We therefore isolated platelets from mice after 7 days of DSS-induced colitis or from untreated control mice and compared HYAL2 protein and hyaluronidase activity. HYAL2 protein levels were an average of 44% lower in platelets from DSS-treated mice compared with control mice (n = 5; P < .05) (Figure 2A-B), and corresponding enzyme activity measurements revealed that hyaluronidase activity was also significantly reduced (n = 5; P < .05) (Figure 2C). Reactive thrombocytosis is a known feature of IBD and DSS-induced colitis,35 and we therefore compared circulating platelet count before and after colitis. Despite the mild thrombocytopenia associated with HYAL2 null mice, we observed an approximate 52% increase in circulating platelet count after 7 days of DSS (Figure 2D). Comparison of serum HA levels revealed a proportional increase in circulating HA during DSS challenge between control and HYAL2-deficient mice (Figure 2E), consistent with defects in HA turnover.

Platelets from mice with DSS-induced colitis have reduced HYAL2 and hyaluronidase activity. Platelets were isolated from untreated control and DSS-treated mice after 7 days. (A) Platelet lysates corresponding to 10 μg of protein were probed for HYAL2 and actin. (B) Densitometry quantification shows that HYAL2 levels are reduced an average of 44% (*P < .05) in platelets from DSS-treated mice compared with control mice. (C) Platelet lysates were compared for their HA-degrading activities by incubation with 60 nM Förster resonance energy transfer–based HA nanoprobes at pH 4.5. Maximum activity was achieved by recombinant hyaluronidase. Platelets from DSS-treated mice exhibit a 60% reduction (*P < .05) in HA-degrading activity compared with platelets from untreated mice. (D) Changes in circulating platelet count before and after DSS-induced colitis (**P < .01). (E) Serum isolated from DSS-treated mice and control mice at 7 days was tested for HA levels (*P < .05).
Platelets from mice with DSS-induced colitis have reduced HYAL2 and hyaluronidase activity. Platelets were isolated from untreated control and DSS-treated mice after 7 days. (A) Platelet lysates corresponding to 10 μg of protein were probed for HYAL2 and actin. (B) Densitometry quantification shows that HYAL2 levels are reduced an average of 44% (*P < .05) in platelets from DSS-treated mice compared with control mice. (C) Platelet lysates were compared for their HA-degrading activities by incubation with 60 nM Förster resonance energy transfer–based HA nanoprobes at pH 4.5. Maximum activity was achieved by recombinant hyaluronidase. Platelets from DSS-treated mice exhibit a 60% reduction (*P < .05) in HA-degrading activity compared with platelets from untreated mice. (D) Changes in circulating platelet count before and after DSS-induced colitis (**P < .01). (E) Serum isolated from DSS-treated mice and control mice at 7 days was tested for HA levels (*P < .05).
Platelet HYAL2 regulates susceptibility of DSS-induced colitis in mice
Because HYAL2 was deficient in platelets from patients with IBD and from DSS-treated mice, we speculated that the interaction between platelets and the endothelium might play a key role in regulating inflammatory progression. To determine whether the enhanced inflammation and tissue destruction seen in HYAL2 KO mice was due to the inability of blood cells to degrade HA, we generated bone marrow (BM) chimeric mice and subjected them to DSS-induced colitis for 7 days. Histologic scoring of distal colon tissue, weight loss, and the DAI revealed that reconstitution of HYAL2+/+ mice with HYAL2−/− BM increased susceptibility to DSS-induced colitis compared with mice reconstituted with HYAL2+/+ BM as a control (Figure 3; supplemental Figure 1B). Key parameters, including submucosal swelling, epithelial erosion, and immune cell infiltration (supplemental Figure 3), closely recapitulated our previous observations of HYAL2 null mice (Figure 1). Control mice reconstituted with HYAL2−/− BM exhibited normal blood cell counts (supplemental Table 2) except for the expected mild anemia and thrombocytopenia associated with loss of HYAL2 in hematopoietic cells.36,37
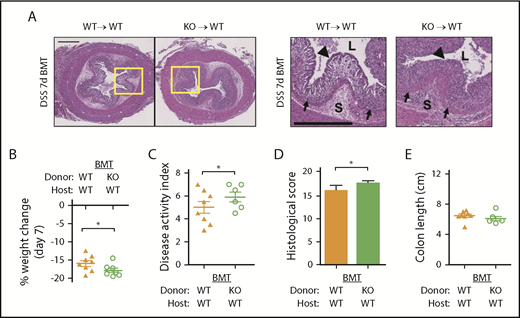
Loss of HYAL2 in hematopoietic cells exacerbates DSS-induced colitis. Bone marrow transplant (BMT) chimeras were generated by using WT (HYAL2+/+) and KO (HYAL2−/−) mice as host and BM donor, respectively. (A) Cross-sections of distal colon collected from mice after 7 days of 2.5% DSS. Hematoxylin-eosin staining reveals the cellular and structural changes in the intestine, including inflammatory leukocyte infiltration. Boxed regions are enlarged in adjacent panels. Scale bars indicate 500 μm. L denotes the intestinal lumen, triangles indicate the epithelial layer, S denotes the submucosa, and arrows indicate the muscularis mucosae. (B) Body weight was measured on day 7 and compared with starting weight. (C) Mice were scored for outward signs of disease, including weight loss, hunched posture, ruffled fur, bloody stools, and rectal prolapse. (D) Distal colon sections from mice after 7 days of DSS were scored for pathologic changes, including erosion of the epithelial layer, leukocyte infiltration, submucosal swelling, muscularis mucosae hyperplasia, and increased vascularization. (E) Colons from mice euthanized after 7 days of DSS were measured for distance (centimeters) from the distal end of the cecum to the rectum. Symbols indicate genotypes and treatment: (▲) WT mice receiving WT BM, n = 8; and (○) WT mice receiving HYAL2 KO BM, n = 7. Data are reported as mean ± SEM. *P < .05.
Loss of HYAL2 in hematopoietic cells exacerbates DSS-induced colitis. Bone marrow transplant (BMT) chimeras were generated by using WT (HYAL2+/+) and KO (HYAL2−/−) mice as host and BM donor, respectively. (A) Cross-sections of distal colon collected from mice after 7 days of 2.5% DSS. Hematoxylin-eosin staining reveals the cellular and structural changes in the intestine, including inflammatory leukocyte infiltration. Boxed regions are enlarged in adjacent panels. Scale bars indicate 500 μm. L denotes the intestinal lumen, triangles indicate the epithelial layer, S denotes the submucosa, and arrows indicate the muscularis mucosae. (B) Body weight was measured on day 7 and compared with starting weight. (C) Mice were scored for outward signs of disease, including weight loss, hunched posture, ruffled fur, bloody stools, and rectal prolapse. (D) Distal colon sections from mice after 7 days of DSS were scored for pathologic changes, including erosion of the epithelial layer, leukocyte infiltration, submucosal swelling, muscularis mucosae hyperplasia, and increased vascularization. (E) Colons from mice euthanized after 7 days of DSS were measured for distance (centimeters) from the distal end of the cecum to the rectum. Symbols indicate genotypes and treatment: (▲) WT mice receiving WT BM, n = 8; and (○) WT mice receiving HYAL2 KO BM, n = 7. Data are reported as mean ± SEM. *P < .05.
We therefore sought to determine whether HYAL2 null mice could be rescued by transplant of control BM. Unexpectedly, irradiation was lethal for 23 of 30 healthy, untreated KO mice (supplemental Figure 4A). Although the few surviving HYAL2−/− mice reconstituted with HYAL2+/+ BM appeared to be less severely affected by DSS (supplemental Figure 3; supplemental Figure 4B-C), the low number of survivors made interpretation underpowered in this model and incomplete.
We therefore directly examined the role of platelet HYAL2 in DSS-induced colitis by adoptive transfer of platelets into KO mice (Figure 4). Because HYAL2 KO mice do not exhibit a consumptive thrombocytopenia,20 we were able to adjust platelet counts to normal levels by platelet transfusion. In the case of KO mice receiving WT platelets, ∼70% of circulating platelets contained HYAL2 at the onset of colitis. Survival of adoptively transferred platelets was measured by using in vivo biotinylation, and no difference in platelet life span was observed (data not shown). Transfused mice were evaluated for weight loss, the DAI, and histologic indices of disease over 7 days of DSS-induced colitis. Mice receiving adoptive transfer of HYAL2-containing platelets exhibited protection from weight loss, less severe signs of disease activity, and histologic evidence of colon sparing compared with mice receiving HYAL2-deficient platelets as a control (supplemental Figure 1C; supplemental Figure 3). Platelets from HYAL2-deficient mice have been shown to activate and spread normally,36 and we further evaluated these platelets for α granule release and agonist response; no significant differences were observed compared with control platelets (supplemental Figure 5).
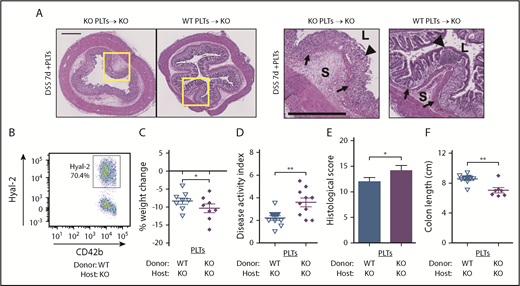
Platelet HYAL2 mediates susceptibility to colitis. Mice received adoptive transfer of 8 × 108 WT or KO platelets as indicated at the onset of DSS treatment. (A) Cross-sections of distal colon collected from mice after 7 days of 2.5% DSS. Hematoxylin-eosin staining reveals the cellular and structural changes in the intestine, including inflammatory leukocyte infiltration. Boxed regions are enlarged in adjacent panels. Scale bars indicate 500 μm. L denotes the intestinal lumen, triangles indicate the epithelial layer, S denotes the submucosa, and arrows indicate the muscularis mucosae. (B) The percentage of platelets expressing HYAL2 in KO mice receiving WT platelets assessed by using flow cytometry. (C) Body weight was measured on day 7 and compared with starting weight. (D) Mice were scored for outward signs of disease, including weight loss, hunched posture, ruffled fur, bloody stools, and rectal prolapse. (E) Distal colon sections from mice after 7 days of DSS were scored for pathologic changes, including erosion of the epithelial layer, leukocyte infiltration, submucosal swelling, muscularis mucosae hyperplasia, and increased vascularization. (F) Colons from mice euthanized after 7 days of DSS were measured for distance (centimeters) from the distal end of the cecum to the rectum. Symbols indicate genotypes and treatment: (∇) KO mice transfused with WT platelets, n = 8; and (♦) KO mice transfused with KO platelets, n = 8. Data are reported as mean ± SEM. *P < .05; **P < .01.
Platelet HYAL2 mediates susceptibility to colitis. Mice received adoptive transfer of 8 × 108 WT or KO platelets as indicated at the onset of DSS treatment. (A) Cross-sections of distal colon collected from mice after 7 days of 2.5% DSS. Hematoxylin-eosin staining reveals the cellular and structural changes in the intestine, including inflammatory leukocyte infiltration. Boxed regions are enlarged in adjacent panels. Scale bars indicate 500 μm. L denotes the intestinal lumen, triangles indicate the epithelial layer, S denotes the submucosa, and arrows indicate the muscularis mucosae. (B) The percentage of platelets expressing HYAL2 in KO mice receiving WT platelets assessed by using flow cytometry. (C) Body weight was measured on day 7 and compared with starting weight. (D) Mice were scored for outward signs of disease, including weight loss, hunched posture, ruffled fur, bloody stools, and rectal prolapse. (E) Distal colon sections from mice after 7 days of DSS were scored for pathologic changes, including erosion of the epithelial layer, leukocyte infiltration, submucosal swelling, muscularis mucosae hyperplasia, and increased vascularization. (F) Colons from mice euthanized after 7 days of DSS were measured for distance (centimeters) from the distal end of the cecum to the rectum. Symbols indicate genotypes and treatment: (∇) KO mice transfused with WT platelets, n = 8; and (♦) KO mice transfused with KO platelets, n = 8. Data are reported as mean ± SEM. *P < .05; **P < .01.
Platelet HYAL2 deficiency exacerbates inflammation in mice with DSS-induced colitis
Previous studies from our laboratory have shown that TNF-α is a potent inducer of HA on endothelial surfaces by regulation of the enzyme hyaluronan synthase 3 (HAS3),19 and genetic knock-out of HAS3 reduces inflammation in DSS-treated mice.26 We therefore analyzed serum TNF-α and interleukin-6 (IL-6) in control and DSS-treated mice (Figure 5A-B). No difference was observed in WT or KO mice at baseline, but at day 7, levels of both TNF-α and IL-6 were significantly increased in HYAL2 KO mice compared with WT control mice (P < .01). Transfusion of HYAL2 KO mice with WT platelets resulted in a significant reduction in TNF-α and IL-6 levels compared with KO alone (P < .01) or KO mice transfused with KO platelets (P < .001).

Platelet HYAL2 regulates serum inflammatory cytokines and inflammatory leukocyte infiltration in mice with DSS-induced colitis. Serum isolated from DSS-treated and control mice at 7 days was tested for (A) TNF-α and (B) IL-6 by using an enzyme-linked immunosorbent assay. (C) F4/80-positive area expressed as the percentage of total area. (D) Ly6G-positive area expressed as the percentage of total area. (E) MPO activity was measured in distal colon lysates of untreated and DSS-treated mice with or without platelet transfusion at 7 days. Data are reported as mean ± SEM; n = 10 mice per group. *P < .05; **P < .01; ***P < .001.
Platelet HYAL2 regulates serum inflammatory cytokines and inflammatory leukocyte infiltration in mice with DSS-induced colitis. Serum isolated from DSS-treated and control mice at 7 days was tested for (A) TNF-α and (B) IL-6 by using an enzyme-linked immunosorbent assay. (C) F4/80-positive area expressed as the percentage of total area. (D) Ly6G-positive area expressed as the percentage of total area. (E) MPO activity was measured in distal colon lysates of untreated and DSS-treated mice with or without platelet transfusion at 7 days. Data are reported as mean ± SEM; n = 10 mice per group. *P < .05; **P < .01; ***P < .001.
Macrophage infiltration is increased in DSS-treated mice with HYAL2-deficient platelets
HA accumulation begins in the microvasculature in DSS-induced colitis and corresponds with dense regions of leukocyte infiltration in the inflamed crypts and submucosa of patients with IBD.27,32 We examined immune cell infiltration by measuring F4/80+ macrophages and Ly6G+ neutrophils present in distal colon tissue sections from control and DSS-treated mice after 7 days (supplemental Figure 6). Quantifiable regions of infiltration in the mucosa and submucosa of WT and HYAL2 KO mice were significantly increased in KO mice compared with control mice (P < .01) (Figure 5C-D). Transfusion of KO mice with WT platelets led to a 45% reduction (P < .05) in macrophage and neutrophil staining compared with either KO mice alone or mice receiving KO platelets.
Myeloperoxidase (MPO) activity was next measured as a marker of neutrophils and macrophages (Figure 5E). We found no statistically observable differences in MPO activity in colon tissue from untreated WT or HYAL2 KO mice; however, consistent with the magnitude of inflammation after 7 days of DSS, MPO activity was increased in HYAL2 KO mice compared with control mice (P < .01). Transfusion of WT platelets into KO mice reduced MPO activity compared with KO mice alone (P < .05) or compared with mice receiving KO platelets (P < .05). Together, these data support a role for platelet HYAL2 regulating inflammatory cell infiltration in DSS-induced colitis.
Deposition of leukocyte-adhesive HA-HC is increased in HYAL2-deficient mice
In addition to its role amplifying HA production through HAS3, TNF-α stimulates production of the enzyme TNF-stimulated gene 6, which modifies HA into HA-HC and enhances leukocyte recruitment.38 We measured submucosal HA-HC deposition and found a striking increase in HA-HC in HYAL2 KO mice compared with WT mice (Figure 6A). Quantitative analysis of tissue HA revealed that total HA levels increased in response to DSS in both control and HYAL2 KO mice, but transfusion of platelets did not affect total HA levels within colon tissue (Figure 6B). However, KO mice transfused with WT platelets had a dramatic decrease in HA-HC deposition compared with KO alone or mice receiving KO platelets (Figure 6C). This finding is consistent with the reduction of TNF-α levels in serum (Figure 5A) and abrogation of leukocyte infiltration into the tissue of HYAL2 null mice transfused with WT platelets (Figure 5C-D).

Microvascular permeability is increased in mice with DSS-induced colitis with HYAL2-deficient platelets. (A) Distal colon sections from mice treated with 2.5% DSS for 7 days with or without transfusion of platelets were evaluated for HA-HC by immunostaining for HA (green) and the HCs of IαI (red) and counterstained with 4′,6-diamidino-2-phenylindole for nuclei. L denotes the lumen. Lower panels show the submucosal regions at higher magnification. Images are representative of at least 8 mice. (B) Measurement of HA levels in colon tissue from control, DSS-treated mice, and DSS-treated mice receiving platelets. (C) Quantification of colocalization of HA with HCs of IαI as a measurement of HA-HC deposition. (D) Measurement of EB extracted from distal colon tissue of mice treated with or without 2.5% DSS. Data are reported as mean ± SEM; n = 10 mice per group. *P < .05; **P < .01; ***P < .001. Imaging, detection, and software details: TCS SP5 II confocal/multiphoton high-speed upright microscope, HCX PL APO 40X/1.25NA oil immersion objective, HyD system detector, and LAS AF software (all Leica Biosystems). Pearson’s correlation coefficients were obtained by analyzing individual images (layers) of the Z-stack with Image-Pro Plus software.
Microvascular permeability is increased in mice with DSS-induced colitis with HYAL2-deficient platelets. (A) Distal colon sections from mice treated with 2.5% DSS for 7 days with or without transfusion of platelets were evaluated for HA-HC by immunostaining for HA (green) and the HCs of IαI (red) and counterstained with 4′,6-diamidino-2-phenylindole for nuclei. L denotes the lumen. Lower panels show the submucosal regions at higher magnification. Images are representative of at least 8 mice. (B) Measurement of HA levels in colon tissue from control, DSS-treated mice, and DSS-treated mice receiving platelets. (C) Quantification of colocalization of HA with HCs of IαI as a measurement of HA-HC deposition. (D) Measurement of EB extracted from distal colon tissue of mice treated with or without 2.5% DSS. Data are reported as mean ± SEM; n = 10 mice per group. *P < .05; **P < .01; ***P < .001. Imaging, detection, and software details: TCS SP5 II confocal/multiphoton high-speed upright microscope, HCX PL APO 40X/1.25NA oil immersion objective, HyD system detector, and LAS AF software (all Leica Biosystems). Pearson’s correlation coefficients were obtained by analyzing individual images (layers) of the Z-stack with Image-Pro Plus software.
The deposition of HA-HC suggested that the microvasculature of the colon was permeable, allowing serum, which contains IαI, to leak into inflamed intestinal tissue. We examined vascular permeability of the colon by i.v. injection of Evans blue dye (EB) and measured leakage into the colon. Although there was no difference in unchallenged mice, after 7 days of treatment with DSS, HYAL2 KO mice displayed increased EB in colon tissue compared with that of control mice (Figure 6D), suggesting that the endothelium was compromised. Transfusion of control platelets into KO mice at the onset of colitis significantly reduced leakage of EB into the inflamed colon compared with KO mice alone or mice receiving KO platelets.
Platelet Hyal-2 regulates leukocyte trans-endothelial migration
A critical function of the endothelium during inflammation is to regulate the migration of leukocytes to sites of inflammation. HA accumulation within the colonic microvasculature of mice occurs as early as 3 days after DSS treatment, well before inflammatory infiltrate can be observed, where it acts to promote leukocyte recruitment,26,27 and platelets can cleave HA from the surface of cells in an HYAL2-dependent fashion.12,19 Within the microvasculature of the colon, HA modified with HCs was observed in patients with IBD compared with non-IBD control subjects and also within vessels of mice subjected to DSS-induced colitis (Figure 7A; supplemental Figure 7A). To determine whether the increase in vessel HA corresponded with changes in HYAL2, we examined cultured endothelial cells and found that messenger RNA, HYAL2 protein, and activity were deficient in HIMECs from patients with IBD compared with those from non-IBD control subjects (Figure 7B-C; supplemental Figure 7B-C). We next questioned whether TNF-α, which increases HA synthesis in HIMECs, also regulates HYAL2. HIMECs treated with TNF-α showed consistent decreases in HYAL2 protein, messenger RNA, and activity levels (Figure 7D; supplemental Figure 7D-E). These analyses reveal that HYAL2 protein levels are consistent through passage in HIMECs and are responsive to TNF-α.
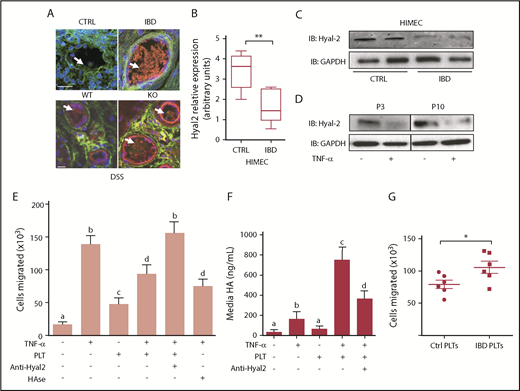
Platelet HYAL2 regulates trans-endothelial PBMC migration. (A) Representative microvessels present in colon tissues isolated from non-IBD control subjects, patients with IBD, and WT or HYAL2 KO mice subjected to DSS-induced colitis were evaluated for HA-HC by immunostaining for HA (green) and the HCs of IαI (red) and counterstained with 4′,6-diamidino-2-phenylindole for nuclei. Arrows indicate HA glycocalyx structures extending from the vessel surface. (B) Quantitative real-time polymerase chain reaction analysis of HYAL2 expression of HIMECs isolated from non-IBD and IBD patient surgical specimens (n = 8 each; **P < .01). (C) Representative immunoblots analyzing HYAL2 levels in HIMECs isolated from non-IBD and IBD patient surgical specimens (n = 8 each). Lysates were normalized to total protein (25 µg), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (D) HIMECs were cultured with or without TNF‐α for 16 hours and evaluated for HYAL2 by using immunoblot over multiple passages. (E) HIMECs were seeded on permeable supports (3 μm pore size) placed into a 24-well plate, and grown to confluence. HIMECs were treated with or without TNF‐α for 16 hours at 37°C to promote leukocyte-adhesive HA-HC formation. Following activation, HIMECs were incubated in the presence or absence of nonactivated freshly isolated human platelets (100 × 106 per well) for 1 hour at 37°C. In some experiments, HIMECs were treated with Streptomyces hyaluronidase or with platelets preincubated with an HYAL2-blocking antibody as indicated. CCL5 (100 ng/mL) was added to the bottom well, and Calcein AM–labeled PBMCs (1 × 106 per well) were added to the upper chamber. (F) HIMECs were cultured with or without TNF-α to induce HA-HC formation on the cell surface. Cultures were then washed and incubated in the presence or absence of platelets as indicated. HA released into the media was measured by using an enzyme-linked immunosorbent assay–like method, and the data represent 3 independent experiments. (G) TNF-α–stimulated HIMECs were pretreated with platelets isolated from IBD platelets or non-IBD control subjects before PBMC trans-endothelial migration. Data are reported as mean ± SEM; n = 4 independent experiments of at least 6 patients each. Values with different alphabetical superscripts are significantly different from each other (P < .05); *P < .05. Image, detection, and software details: TCS SP5 II confocal/multi-photon high-speed upright microscope, HCX PL APO ×40/1.25NA oil immersion objective, HyD system detector, and LAS AF software (all Leica Biosystems). Scale bar: 25 µm. Data acquisition details: Calcein AM–labeled PBMCs were detected in the lower wells with an automated Leica DM inverted microscope at 20× magnification set to well-scan mode. PBMCs were enumerated on the basis of fluorescence and size by using Image-Pro Plus acquisition software.
Platelet HYAL2 regulates trans-endothelial PBMC migration. (A) Representative microvessels present in colon tissues isolated from non-IBD control subjects, patients with IBD, and WT or HYAL2 KO mice subjected to DSS-induced colitis were evaluated for HA-HC by immunostaining for HA (green) and the HCs of IαI (red) and counterstained with 4′,6-diamidino-2-phenylindole for nuclei. Arrows indicate HA glycocalyx structures extending from the vessel surface. (B) Quantitative real-time polymerase chain reaction analysis of HYAL2 expression of HIMECs isolated from non-IBD and IBD patient surgical specimens (n = 8 each; **P < .01). (C) Representative immunoblots analyzing HYAL2 levels in HIMECs isolated from non-IBD and IBD patient surgical specimens (n = 8 each). Lysates were normalized to total protein (25 µg), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control. (D) HIMECs were cultured with or without TNF‐α for 16 hours and evaluated for HYAL2 by using immunoblot over multiple passages. (E) HIMECs were seeded on permeable supports (3 μm pore size) placed into a 24-well plate, and grown to confluence. HIMECs were treated with or without TNF‐α for 16 hours at 37°C to promote leukocyte-adhesive HA-HC formation. Following activation, HIMECs were incubated in the presence or absence of nonactivated freshly isolated human platelets (100 × 106 per well) for 1 hour at 37°C. In some experiments, HIMECs were treated with Streptomyces hyaluronidase or with platelets preincubated with an HYAL2-blocking antibody as indicated. CCL5 (100 ng/mL) was added to the bottom well, and Calcein AM–labeled PBMCs (1 × 106 per well) were added to the upper chamber. (F) HIMECs were cultured with or without TNF-α to induce HA-HC formation on the cell surface. Cultures were then washed and incubated in the presence or absence of platelets as indicated. HA released into the media was measured by using an enzyme-linked immunosorbent assay–like method, and the data represent 3 independent experiments. (G) TNF-α–stimulated HIMECs were pretreated with platelets isolated from IBD platelets or non-IBD control subjects before PBMC trans-endothelial migration. Data are reported as mean ± SEM; n = 4 independent experiments of at least 6 patients each. Values with different alphabetical superscripts are significantly different from each other (P < .05); *P < .05. Image, detection, and software details: TCS SP5 II confocal/multi-photon high-speed upright microscope, HCX PL APO ×40/1.25NA oil immersion objective, HyD system detector, and LAS AF software (all Leica Biosystems). Scale bar: 25 µm. Data acquisition details: Calcein AM–labeled PBMCs were detected in the lower wells with an automated Leica DM inverted microscope at 20× magnification set to well-scan mode. PBMCs were enumerated on the basis of fluorescence and size by using Image-Pro Plus acquisition software.
Based on these findings, we hypothesized that platelet HYAL2 regulates inflammatory progression by degrading HA on the surface of inflamed vessels to limit leukocyte emigration. To test this hypothesis, HIMECs were grown on Transwell filters without or with TNF-α (10 ng/mL) in the presence of human serum to induce formation of leukocyte-adhesive HA-HC. After activation, HIMECs were incubated in the presence or absence of nonactivated freshly isolated platelets (100 × 106 per well) for 1 hour followed by gentle washing. An HIMEC-independent chemotactic factor (CCL5; 100 ng/mL) was added to the bottom well before the addition of human peripheral blood mononuclear cells (PBMCs) to the upper chamber, and transmigration was measured by analyzing the number of PBMCs present in the bottom chamber after 3 hours (Figure 7E). Stimulation of HIMECs with TNF-α led to an increase in transmigration (P < .001). Preincubation of HIMECs with platelets also increased migration but to a lesser extent (P < .05) compared with untreated control cells. Preincubation of TNF-stimulated HIMECs with platelets decreased migration an average of 35% (P < .001) compared with TNF-stimulated HIMECs without platelet treatment.
To test whether the observed decrease in migration was due to the activity of HYAL2, we treated platelets with an HYAL2-neutralizing antibody or with nonspecific control immunoglobulin G and removed excess antibody by centrifugation. PBMC transmigration across TNF-stimulated HIMEC cultures preincubated with the HYAL2-blocked platelets was equivalent to leukocyte migration across TNF-stimulated HIMECs alone. Control immunoglobulin G–treated platelets did not abrogate PBMC migration and were as effective as untreated platelets (data not shown). These findings indicate that platelets can reduce PBMC transmigration across an activated HIMEC monolayer through an HYAL2-dependent mechanism. We further assayed HA release into the media from replicate cultures of HIMECs and observed that incubation of TNF-stimulated HIMECs with platelets results in increased media HA (P < .001), which can be significantly inhibited by pretreating platelets with an HYAL2-neutralizing antibody (Figure 7F). This scenario suggests that platelets can limit PBMC transmigration by releasing HA from the surface of activated HIMECs.
We previously reported that platelets isolated from patients with IBD exhibit a 45% reduction in HYAL2 protein levels and a corresponding decrease in hyaluronidase activity.12 To determine whether this deficiency affects the ability of platelets to regulate leukocyte transmigration, we compared unstimulated, freshly isolated platelets from healthy donor subjects and from patients with IBD (at least 6 each) using the same trans-endothelial migration assay. As shown in Figure 7G, preincubation of TNF-stimulated HIMECs with platelets from patients with IBD resulted in substantially increased (average 130% of control; P < .05) PBMC migration compared with HIMECs treated with platelets from healthy donor subjects. This finding suggests that the platelet HYAL2-dependent mechanism is disrupted in the IBD patient population and may result in sustained expression of leukocyte-adhesive HA-HC by the microvasculature, impairing the resolution of inflammation and promoting further tissue damage.
Discussion
Using a combination of patient tissues, animal models, and patient cells, we provide evidence that platelets regulate the early stages of inflammation by limiting leukocyte extravasation via HYAL2-dependent degradation of endothelial HA. We show that loss of HYAL2 in mice exacerbated disease activity in the DSS-induced experimental colitis model. Importantly, we found that increased susceptibility to DSS-induced colitis in the HYAL2-KO background can be significantly abrogated by transfusion of platelets expressing HYAL2 at the initiation of colitis, and that this effect occurs in the microvasculature. We also show for the first time that HYAL2 is deficient in cultured intestinal endothelial cells isolated from patients with IBD and that mice subjected to DSS-induced colitis have diminished platelet-HYAL2 and hyaluronidase activity similar to the IBD patient platelets. We found that this protective mechanism in platelets is HYAL2 dependent and impaired in platelets isolated from patients with IBD. Finally, these animal model data reveal that platelet HYAL2 is a protective mechanism controlling colitis, and that HYAL2-competent platelet supplementation for patients with IBD presents an unanticipated opportunity to investigate a new arm of treatment to add to the current modalities.
A growing number of studies underscore the importance of nonimmune cells such as mesenchymal, endothelial, and platelets as key players in the progression of IBD inflammatory cascade.1,4 The results of the current investigation highlight the interplay between platelets and the microvasculature during the early stages of colitis. HA accumulates in small vessels at the early stages of pathologic changes in the inflamed distal colon before inflammatory cell infiltration,27 and HA supports leukocyte and platelet adhesion, as shown in vitro.32,39,40 Here, we show that the HA-degrading enzyme HYAL2 is altered in microvascular endothelial cells isolated from patients with IBD at the messenger RNA level, with a corresponding deficiency in HYAL2 protein and HA degradation activity (Figure 7B-C; supplemental Figure 7B-C). Our data reveal that in response to TNF-α, HYAL2 becomes downregulated (Figure 7D; supplemental Figure 7D-E), supporting previous observations that conditions which promote production of proinflammatory HA may also suppress hyaluronidase activity.41 We previously reported that HIMECs isolated from patients with IBD overproduce leukocyte-adhesive HA compared with HIMECs isolated from healthy control subjects.19 This observation is consistent with reports that HYAL2 acts as a negative regulator of the glycocalyx42 such that the reduced HYAL2 in IBD HIMECs would result in increased cell surface HA. Unstimulated HIMECs isolated from patients with IBD support greater leukocyte adhesion compared with HIMECs from non-IBD control subjects,43 and increased HA may contribute.
The ability of inflammation-associated modification of ECM (ie, HA-HC) to enhance leukocyte recruitment is a general mechanism relevant in many disease states beyond IBD.24-29 HA via its interaction with CD44 plays a crucial for leukocyte infiltration into inflammatory sites, and multiple studies show that disruption of this interaction by genetic ablation,24,44 CD44 antibody blockade,29,39,45,46 or digestion of cell surface HA with hyaluronidase28,47 is capable of abolishing HA-dependent cell adhesion. Although other HA receptors may also play a role in this process, to date, CD44 is the most extensively studied. Consistent with our studies, early administration of recombinant human hyaluronidase before signs of disease activity occur has been shown to inhibit reactive adipogenesis, inflammation, and tissue injury in a mouse DSS model.48 To the best of our knowledge, our report is the first to show that platelets can regulate this process and also that circulating cells rather than a soluble enzyme are mediators.
Our data raise several intriguing questions regarding the role of HA in the initiation and perpetuation of inflammation. We previously recognized that disruption of HA synthesis by HAS3 significantly reduces the inflammatory response in a murine model of colitis.26 We now understand that disruption of HA turnover in mice by either systemic deletion of HYAL2 or generation of HYAL2 null BM chimeras increases susceptibility to DSS-induced colitis (Figure 5). The finding that HYAL2 is deficient in IBD platelets and HIMECs, and in mouse platelets during experimental colitis, was unexpected. HAS3 is induced by TNF-α,26 and in HIMECs, it also leads to downregulation of HYAL2 (Figure 7D; supplemental Figure 7D-E); however, the data available for HYAL2 in other cell types are contradictory41,49,50 and may reflect cell type–specific responses. Our data suggest that HAS3 and HYAL2 may be regulated in a temporal fashion in response to inflammation, such that HA functions to recruit immune cells and HYAL2 acts to limit inflammation to enable resolution.
Although our data describe an early, protective role for platelets in colitis, it is likely that platelets exacerbate inflammation later in the disease. Many proinflammatory functions of platelets have been noted in IBD, and HA fragments released from the surface of inflamed cells can activate monocytes, endothelial cells, and smooth muscle cells.19 Whether those fragments reach concentrations that can signal in circulation or locally at the site of their generation is not known. At extravascular sites of platelet accumulation, such as the inflamed submucosa, it is plausible that both platelets and HA fragments reach concentrations capable of driving a self-perpetuating loop that contributes to inflammatory disease.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Gail West for assistance with cell isolation, Claudio Fiocchi for helpful conversations, and Satya Kurada for assistance with IBD patient samples.
This work was financially supported by the National Institutes of Health (K99HL135265) (A.C.P.) and the Programs of Excellence in Glycosciences (HL107147) from the National Heart, Lung, and Blood Institute (C.A.d.l.M).
Authorship
Contribution: A.C.P. designed the research, performed experiments, analyzed results, and wrote the paper; D.R.O. performed experiments and analyzed results; S.P.K. designed the research, performed experiments, and analyzed results; A.Z. performed experiments; B.F. provided the mice; and C.A.d.l.M. designed the research, analyzed results, and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carol A. de la Motte, Cleveland Clinic Lerner Research Institute, Department of Inflammation and Immunity, NC22, 9500 Euclid Ave, Cleveland, OH 44195; e-mail: delamoc@ccf.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal