

Key Points
Recombinant WT APC or signaling-selective APC variant therapy reduces murine cGVHD pathology.
APC’s biased PAR1-dependent signaling due to Arg46 cleavage in PAR1 on donor T cells is required for APC’s in vivo benefits in cGVHD.

Abstract
Soluble thrombomodulin plasma concentrations are elevated in steroid-resistant graft-versus-host disease (GVHD), implying endothelial hypofunctioning for thrombomodulin-dependent generation of activated protein C’s (APC) anticoagulant, anti-inflammatory, and antiapoptotic functions. Recombinant thrombomodulin or APC administration decreases acute GVHD, manifested by intense inflammation and tissue destruction. Here, we administered recombinant murine wild-type (WT) APC to mice with established chronic GVHD (cGVHD), a less-inflammatory autoimmune-like disease. WT APC normalized bronchiolitis obliterans–induced pulmonary dysfunction. Signaling-selective APC variants (3A-APC [APC with lysine 191-193 replaced with 3 alanines] or 5A-APC [APC with lysine 191-193 replaced with 3 alanines and arginine 229/230 replaced with 2 alanines]) with normal cytoprotective properties, but greatly reduced anticoagulant activity, provided similar results. Mechanistically, WT APC and signaling-selective variants reduced T follicular helper cells, germinal center formation, immunoglobulin, and collagen deposition. WT APC can potentially cleave protease-activated receptor 1 (PAR1) at Arg41 or Arg46, the latter causing anti-inflammatory signaling. cGVHD was reduced in recipients of T cells from WT PAR1 or mutated Gln41-PAR1 donors but not from mutated Gln46-PAR1 donors. These data implicate donor T-cell APC-induced noncanonical cleavage at Arg46-PAR1, which is known to confer cytoprotective and anti-inflammatory activities. Together, these data indicate that APC anticoagulant activity is dispensable, whereas anti-inflammatory signaling and cytoprotective cell signaling by APC are essential. Because a phase 2 ischemic stroke clinical trial did not raise any safety issues for 3A-APC treatment, our studies provide a foundational platform for testing in clinical cGVHD therapy.
Introduction
Chronic graft-versus-host disease (cGVHD) is a major cause of morbidity and mortality following allogeneic hematopoietic cell transplantation (allo-HCT).1-3 Activated protein C (APC) is a plasma serine protease that cleaves protease-activated receptor 1 (PAR1) at canonical Arg41 or the noncanonical Arg46 site; the latter triggers anti-inflammatory, antiapoptotic, and broad cytoprotective activities.4 APC’s cell signaling and anticoagulant activities are potentially advantageous in treating allo-HCT patients prone to tissue injury and thrombotic events.
APC has been implicated in acute graft-versus-host disease (aGVHD) because of the decreased endothelial thrombomodulin needed to generate APC, especially in steroid-resistant aGVHD; conversely, recombinant thrombomodulin induced murine aGVHD amelioration.5,6 APC can restrain allogeneic T-cell activation by stimulating regulatory T cells via PAR2/PAR3 homodimers.5 In contrast to inflammatory tissue-destructive aGVHD processes, murine and human cGVHD may manifest as a primarily fibrotic disease associated with autoantibodies or alloantibodies. To facilitate isotype-switched high-affinity antibodies, T follicular helper cells (TFHs) engage with germinal center (GC) B cells. In a multiorgan system cGVHD model with bronchiolitis obliterans (BO) (cGVHD/BO), TFHs, GC B cells, and GC size and number are increased, whereas suppressive T follicular regulatory cells (TFRs) are decreased.7 This imbalanced TFR/TFH ratio is permissive of immunoglobulin (Ig) production, resulting in IgG2c deposition in lung, liver, and colon, as well as fibrosis due to Fc receptor-bearing transforming growth factor-β–releasing macrophages.7-10
To discern whether APC’s antithrombotic or cytoprotective and tissue-regenerative properties were critical in treating cGVHD mice, mice experiencing ongoing cGVHD/BO were treated with wild-type (WT) APC or signaling-selective variants with greatly reduced anticoagulant activities, but intact cytoprotective activities: 3A-APC (APC with lysine 191-193 replaced with 3 alanines) or 5A-APC (APC with lysine 191-193 replaced with 3 alanines and Arg229/330 replaced with 2 alanines). Alternatively, donor T cells mutated at canonical or noncanonical PAR1 cleavage sites were infused to cause cGVHD. Together, our data show that APC-induced biased signaling involving noncanonical cleavage at PAR1’s Arg46 in donor T cells is essential for cGVHD benefits.
Study design
Mice
C57BL/6 (B6;H2b) mice (National Cancer Institute) and B10.BR (H2k) mice (The Jackson Laboratory) were housed in a pathogen-free facility under University of Minnesota Institutional Animal Care Committee guidelines. Purified donor T cells were isolated from shipped splenocytes from WT littermates or PAR1-genetically modified mice with R41Q or R46Q point mutations.11
cGVHD
B10.BR recipients were given cyclophosphamide (120 mg/kg per day, days −3 and −2), total body irradiation (TBI; 8.3 Gy, day −1), and B6 T-cell–depleted bone marrow (BM; 107 cells), with or without 0.6 to 0.8 × 105 T cells (day 0).8 cGVHD assessments included pulmonary function tests for BO, flow cytometry, immunofluorescent microscopy, Ig and trichrome staining, and quantification.8 Survival of mice with no cGVHD (receiving donor BM alone) or cGVHD (receiving donor BM + T cells) ranged from 86-100% (n = 38 per group).
WT APC and variants
Flow cytometry and tissue analyses
With the exception of sheep red blood cell immunized (SRBC) mice in Figure 1E, all flow cytometry was performed on a Fortessa and included Fixable Viability Dye APC-eF780 (eBioscience). GC B cells were defined as CD19+, CD95 (FAS)(hi), GL7+, TFH cells as CD4+, PD-1+, CXCR5+, FoxP3− and TFR cells as CD4+, PD-1+, CXCR5+, FoxP3+. Reagents from eBioscience (CD19, GL7, PD-1, FoxP3), BD Bioscience (FAS, CD4) and BD Pharmingen (CXCR5) were used. For SRBC immunized mice, flow cytometry analysis was performed on an LSR II and included gating on live cells by exclusion of Ghost Violet GV510 stain; doublet cells were excluded by gating forward scatter height vs forward scatter width, and cells were stained using reagents from eBioscience, BD Biosciences, BioLegend, or Tonbo Biosciences. GC B cells were identified as B220+, FAS+, GL7+, TFHs were identified as CD4+, TCRb+, FoxP3−, PD-1hi, Bcl6+, and TFRs were identified as CD4+, TCRb+, FoxP3+, PD-1hi, Bcl6+.
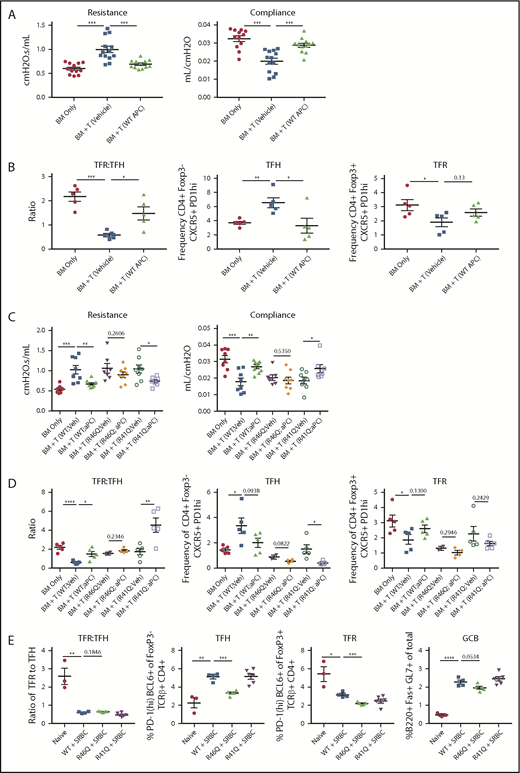
Therapy with WT APC or signaling-selective 5A-APC improves GVHD/BO and requires arginine 46 (R46) in PAR1 expressed on donor T cells. B10.BR (H2k) mice were conditioned with 120 mg/kg cyclophosphamide for 2 days, followed by 830 cGy TBI. On day 0, mice were transplanted with C57BL/6 (H2b) T-cell depleted BM and purified splenic T cells (7 × 104). Mice were allowed to develop disease and were treated daily with 6 µg of recombinant murine WT APC (A-B) or 5A-APC (C-D; abbreviated herein as APC) intraperitoneally from days 28 to 56. (A,C) Pulmonary function tests were performed on day 56 by measuring resistance and compliance after treatment. (B,D) Spleens of BM-only (no cGVHD) or cGVHD mice, treated as indicated, were analyzed on day 56 for the frequency of TFHs, frequency of TFRs, and TFR/TFH ratio. Three (A) or 2 (C) experiments were performed for pulmonary function, and 1 experiment was performed for flow cytometry with 4 or 5 mice analyzed per group (B,D). (E) Sheep red blood cells (SRBCs) were administered intraperitoneally (n = 4-6 mice per group). Seven days later, spleens and sera from naive or SRBC-immunized WT, arg46 (R46Q), or arg41 (R41Q)–mutated mice were analyzed. Data are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Therapy with WT APC or signaling-selective 5A-APC improves GVHD/BO and requires arginine 46 (R46) in PAR1 expressed on donor T cells. B10.BR (H2k) mice were conditioned with 120 mg/kg cyclophosphamide for 2 days, followed by 830 cGy TBI. On day 0, mice were transplanted with C57BL/6 (H2b) T-cell depleted BM and purified splenic T cells (7 × 104). Mice were allowed to develop disease and were treated daily with 6 µg of recombinant murine WT APC (A-B) or 5A-APC (C-D; abbreviated herein as APC) intraperitoneally from days 28 to 56. (A,C) Pulmonary function tests were performed on day 56 by measuring resistance and compliance after treatment. (B,D) Spleens of BM-only (no cGVHD) or cGVHD mice, treated as indicated, were analyzed on day 56 for the frequency of TFHs, frequency of TFRs, and TFR/TFH ratio. Three (A) or 2 (C) experiments were performed for pulmonary function, and 1 experiment was performed for flow cytometry with 4 or 5 mice analyzed per group (B,D). (E) Sheep red blood cells (SRBCs) were administered intraperitoneally (n = 4-6 mice per group). Seven days later, spleens and sera from naive or SRBC-immunized WT, arg46 (R46Q), or arg41 (R41Q)–mutated mice were analyzed. Data are mean ± standard error of the mean. *P < .05, **P < .01, ***P < .001, ****P < .0001.
Statistics
Significance was analyzed using the Student t test, and survival was analyzed using the log-rank test.
Results and discussion
APC signaling is required to treat established cGVHD
In TBI and acute lung injury models, APC provided cytoprotective and anti-inflammatory effects against lethality.4 Efficacy has been observed in murine autoimmunity models,4 including multiple sclerosis, systemic lupus erythematosus, and nonobese diabetes.14-16 These preclinical results, along with APC anticoagulant, anti-inflammatory, and cytoprotective effects,4 prompted our investigation of WT APC as a cGVHD therapeutic. Mice with established cGVHD/BO were given recombinant WT APC on days 28 to 56. In cGVHD/BO mice, heightened resistance and low compliance, indicative of stiff lungs and pulmonary fibrosis, were significantly improved by WT APC to levels equaling or approaching no-cGVHD controls (Figure 1A). Consistent with improved pulmonary function, the TFR/TFH ratio, which is key in controlling the GC response, was significantly increased by APC treatment (Figure 1B); WT APC also increased TFRs, albeit to a slightly lesser extent (P = .13).
To discern between APC anticoagulant activity and cell-signaling actions,4 we infused WT APC or 1 of the 2 signaling-selective APCs (3A-APC or 5A-APC) with greatly reduced anticoagulant, but normal cell-signaling, activities from days 28 to 56 and then measured pulmonary function. Compared with controls, lung function parameters were significantly improved following APC administration (Figure 2A). Pulmonary function in mice receiving WT APC, 3A-APC, or 5A-APC was indistinguishable from one other and from no-cGVHD controls, whereas vehicle-treated mice had BO manifestations. Interleukin-17A (IL-17A) supports murine and human GC formation and cGVHD.17,18 APC ligation of its receptor, endothelial protein C receptor (EPCR), negatively regulates T helper 17 (Th17) cell differentiation,17,18 which is potentially advantageous in cGVHD treatment. cGVHD/BO mice given WT APC, 3A-APC, or 5A-APC each decreased the percentages of Th17-expressing nonregulatory CD4+ T cells and TFHs (P = .07, <.05, and <.05, respectively) (Figure 2B-C). Consistent with lower percentages of IL-17+ T cells and TFHs, GC size and frequency were significantly decreased (Figure 2D). Reflective of decreased GC size, lung IgG deposition was significantly reduced by WT APC, 3A-APC, and 5A-APC (Figure 2E), as reflected by concordant quantitative decreases in collagen deposition and fibrosis (Figure 2F). These data point to APC-induced cell signaling as being required for efficacy in this cGVHD/BO model.
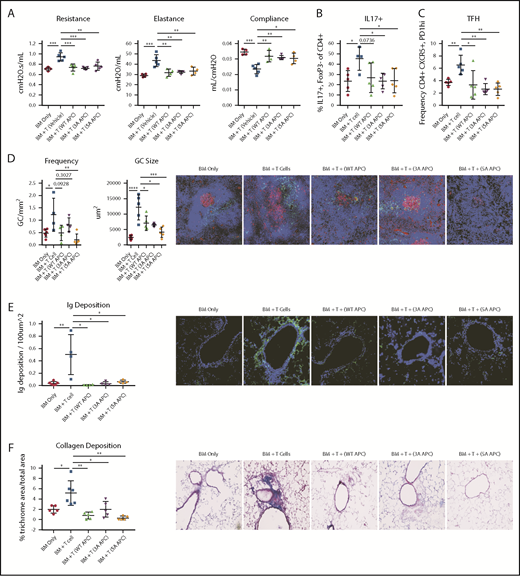
WT or signaling-selective APC-induced therapeutic improvement in cGVHD/BO involves decreased percentages of IL-17 and TFHs associated with reductions in GCs, Ig, and collagen deposition. cGVHD/BO was induced per Figure 1, and mice were given WT APC or signaling-selective 3A-APC or 5A-APC intraperitoneally from days 28 to 56. (A) Pulmonary function test results are shown. Spleens of BM-only (no cGVHD) or cGVHD mice, treated as indicated, were analyzed on day 56 by flow cytometry for IL-17+ T cells (B) and TFHs (C). (D) Immunofluorescent microscopy and quantitative imaging were performed for spleen GC size and frequencies. (E) Lung Ig deposition data using 4′,6-diamidino-2-phenylindole (blue) and anti-IgG-FITC (green) (BD Pharmigen). (F) Lung collagen deposition was analyzed by Masson’s trichrome staining. One experiment is shown, with 4 or 5 mice analyzed per group. Mean values ± standard error of the mean are shown. Original magnification ×200 for panels D-F. *P < .05, **P < .01, ***P < .001, ****P < .0001.
WT or signaling-selective APC-induced therapeutic improvement in cGVHD/BO involves decreased percentages of IL-17 and TFHs associated with reductions in GCs, Ig, and collagen deposition. cGVHD/BO was induced per Figure 1, and mice were given WT APC or signaling-selective 3A-APC or 5A-APC intraperitoneally from days 28 to 56. (A) Pulmonary function test results are shown. Spleens of BM-only (no cGVHD) or cGVHD mice, treated as indicated, were analyzed on day 56 by flow cytometry for IL-17+ T cells (B) and TFHs (C). (D) Immunofluorescent microscopy and quantitative imaging were performed for spleen GC size and frequencies. (E) Lung Ig deposition data using 4′,6-diamidino-2-phenylindole (blue) and anti-IgG-FITC (green) (BD Pharmigen). (F) Lung collagen deposition was analyzed by Masson’s trichrome staining. One experiment is shown, with 4 or 5 mice analyzed per group. Mean values ± standard error of the mean are shown. Original magnification ×200 for panels D-F. *P < .05, **P < .01, ***P < .001, ****P < .0001.
APC-biased signaling is involved in APC-induced improvement of cGVHD
APC exerts biased protective signaling upon PAR cleavage at Arg46 (R46Q) residue, in contrast to thrombin-induced deleterious effects, by cleaving at Arg41 (R41Q).4,19 APC-induced cell signaling due to PAR1 Arg46 cleavage is required for its life-saving efficacy in pneumonia and ischemic stroke.11 Because PAR1 is expressed on murine T cells,4,5 we assessed APC’s requirement for donor T-cell PAR1 Arg46 cleavage in ameliorating cGVHD. Chemoradiotherapy-conditioned B10.BR mice were given B6 T-cell–depleted BM and T cells from WT or PAR1 genetically modified (R41Q or R46Q) donors. Given the comparable efficacy of WT and 5A-APC, cohorts were treated from days 28 to 56 with 5A-APC therapy, avoiding undesirable anticoagulant properties for clinical applications. As anticipated, mice receiving WT T cells and 5A-APC had significant improvement in lung function parameters compared with vehicle-treated cGVHD controls (Figure 1C). Similarly, 5A-APC–treated cGVHD mice given R41Q-PAR1–mutated T cells exhibited significantly improved lung function parameters, in contrast to 5A-APC–treated cGVHD mice given R46Q-PAR1–mutated T cells (Figure 1C). Although vehicle-treated recipients of R41Q or R46Q T cells had lower percentages of TFHs than WT controls, R41Q T cells, but not R46Q T cells, given with APC significantly decreased the percentages of TFHs and dramatically increased TFR/TFH ratios. (Figure 1D). Because T-cell–intrinsic biases may have skewed GC TFR, TFH, and B-cell responses in PAR1-mutated T cells, we immunized naive mice with the potent immunogen, sheep red blood cells, and assessed GC cellular responses. Sheep red blood cell–immunized R41Q-PAR1 mice had higher percentages of TFHs than did immunized R46Q-PAR1 mice, yet APC-mediated cGVHD amelioration was intact in R41Q-PAR1, but not R46Q-PAR1, mice that had lower percentages of TFH and GC B cells (Figure 1C-D). Arg46’s requirement for APC’s cGVHD efficacy strongly supports the hypothesis that PAR1-dependent biased signaling via Arg46 cleavage mediates APC’s in vivo cGVHD benefits, although we cannot exclude that the donor environment contributes to differences. The PAR1 requirement for APC’s cGVHD benefits contrasts sharply with its aGVHD benefits, for which PAR2 and PAR3, but not PAR1, were required.5 In aGVHD, blocking APC binding to EPCR did not abolish APC’s inhibitory effect on T-cell activation,5 consistent with an EPCR-independent T-cell signaling mechanism. In contrast, EPCR is a negative regulator for Th17 pathogenicity, a critical T-cell attribute for cGVHD induction,18 providing a plausible explanation for differences in PAR dependencies based upon alloactivated T effector cells and TFHs. Formal testing of EPCR dependency in APC cGVHD effects will require additional study.
In summary, we identified signaling-selective 3A-APC and its receptor, PAR1, for therapeutic treatment of murine cGVHD/BO that has a clear translational pathway. APC’s reliance upon PAR1’s biased signaling, and not on APC’s anticoagulant properties, could be highly desirable in allo-HCT patients who may have low platelets, coagulopathies, thrombotic microangiopathy, or organ (renal, liver) dysfunction; this leverages a 3A-APC phase 2 ischemic stroke trial that was completed without toxicity.20
For original data, contact Bruce Blazar (blaza001@umn.edu).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported in part by National Institutes of Health, National Cancer Institute grant P01 CA142106 and National Institutes of Health, National Institute of Allergy and Infectious Diseases grant P01 AI056299 (B.R.B.), Leukemia and Lymphoma Society Translational Research grants 6458-15 and 6462-15 (B.R.B.), and National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL052246, P01 HL031950, and R01 HL142975 (J.H.G.).
Authorship
Contribution: R.K.S., R.F., K.P., A.L.G., D.N., J.H.G., and B.R.B. designed experiments; R.K.S., R.F., K.P., M.Z., and A.L.G. performed experiments, discussed results, and edited the manuscript; J.A.F. and X.X. provided essential reagents and mice; and R.K.S., J.H.G., and B.R.B. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare that their institutions have intellectual property related to the discoveries in this article. J.H.G. is a consultant for ZZ Biotech LLC.
The current affiliation for R.K.S. is NantKwest, Inc., San Diego, CA.
Correspondence: John H. Griffin, Department of Molecular Medicine, The Scripps Research Institute, IMM316, 10550 N. Torrey Pines Rd, La Jolla, CA 92037; e-mail: jgriffin@scripps.edu; and Bruce R. Blazar, MMC 109, University of Minnesota, 420 Delaware St SE, Minneapolis, MN 55455; e-mail: blazar001@umn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal