

Adult T cell leukemia / lymphoma is a rare neoplasm with dismal cure rates and poor response to chemotherapy. The genetic basis of refractoriness against conventional agents is not known. We follow one of the largest cohorts of ATLL in the US and recently showed that North American ATLL (NA-ATLL) has a distinct mutational profile from Japanese ATLL (J-ATLL) with a higher incidence of epigenetic mutations (Shah et al, Blood, 2018; 132(14):1507-1518). Here, we analyze the mutational landscape of ATLL clones at diagnosis and at relapse in a pilot study of 7 NA-ATLL patients using deep targeted sequencing of 236 recurrently mutated genes.
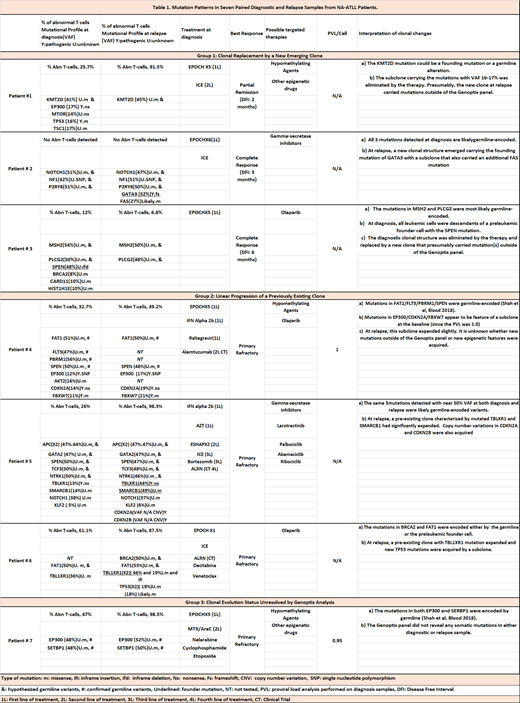
Our goal was to examine clonal evolution patterns that may result as a consequence of the selective pressure of chemotherapy by determining changes in mutational profiles as well as clonal abundance on patients with aggressive ATLL [acute and lymphomatous] (Table 1). All patients were treated with EPOCH (Etoposide, Prednisone, Vincristine, Cyclophosphamide and Doxorubicin) chemotherapy after initial diagnosis except one patient who received interferon therapy.
Distinct patterns of clonal evolution were observed in patients with primary refractory disease when compared to those who relapsed after an initial remission. Group 1 has 3 cases (Pts 1 to 3), all of which experienced early relapse and showed mutation patterns suggestive of clonal replacement by a new emerging clone at relapse. Group 2 has 3 patients (Pts 4-6) that are characterized by worsening of primary refractory disease. Mutation patterns in the before-and-after sample pairs suggest linear progression from a previously existing clone during therapy. The clonal evolution status was not resolved in our third group (Pt#7) indicating that mutations outside the 236 genes evaluated might have been present. Frequent epigenetic mutations were found in both diagnostic and refractory/relapsed samples. Surprisingly, 4 epigenetic mutations (KMT2D, EP300, SPEN, SETBP1) identified in 4 cases were confirmed or suspected of germline origin. This suggests that ancestral genomic differences between the Caribbean and Japanese populations could contribute to the mutational and clinical disparities between NA- and J-ATLL.
Multiple mutations with similar variant allele frequencies (VAF) at the time of relapse were identified, suggesting simultaneous acquisition of several mutations during clonal evolution. Clonal replacement by an emerging clone (Group 1) such as GATA3 (Pt#3) or KMT2D (Pt#1) (either germline or founding mutation) was observed in patients who achieved an initial response. On the other hand, in primary refractory disease (Group 2), relapse was driven by dominant TBL1XR1 mutations in 2 out of the 3 cases either alone (Pt#6) or in combination with mutated SMARCB1 (Pt#5). The protein encoded by TBL1XR1 is a negative regulator of the nuclear receptor corepressor (NCoR) /histone deacetylase 3 (HDAC3) complex. Therefore, TBL1XR1 inactivation is expected to result in hyper-activity of NCoR/HDAC3, an epigenetic abnormality targetable by HDAC inhibitors. SMARCB1 is also an epigenetic regulator associated with several malignancies and has been targeted with alisertib. A subclonal expansion of EP300 (a histone acetyltransferase) appears to drive relapse in the other case of this group (Pt#4). Therefore, our primary refractory cases show that epigenetic mutations seem to be of utmost importance not only at diagnosis as we have previously shown, but as the driving force behind chemotherapy refractoriness and subsequent relapse.
Conclusion: To our knowledge, this is the first mutation-based clonal evolution study aimed to examine dynamic changes induced by chemotherapy among NA-ATLL patients. Early relapse appears to be driven by the emergence of new mutant clones. On the other hand, primary refractory disease appears to have epigenetic drivers that convey chemotherapy refractoriness and evolve from linear progression of a previously existing clone. Serial mutational testing can provide critical information on clonal architecture and identify actionable molecular vulnerabilities in a cohort without effective therapeutic options and a dismal prognosis.
Sica:Physician's Education Resource (PER): Honoraria. Shah:Physicians' Education Resource: Honoraria. Steidl:Aileron Therapeutics: Consultancy, Research Funding; Stelexis Therapeutics: Equity Ownership, Membership on an entity's Board of Directors or advisory committees, Other: Scientific Co-Founder; Pieries Pharmaceuticals: Consultancy; BayerHealthcare: Consultancy, Research Funding; Novartis: Consultancy, Research Funding; GlaxoSmithKline: Research Funding; Celgene: Consultancy. Verma:Stelexis: Equity Ownership, Honoraria; Acceleron: Honoraria; Celgene: Honoraria; BMS: Research Funding; Janssen: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal