

Whole genome sequencing (WGS) studies have started to reveal the critical role of structural variants (SVs) in multiple myeloma (MM) pathogenesis and evolution. We have recently revealed the existence of three main classes of complex events in 30 MM patients: chromothripsis, chromoplexy and templated insertions (Maura F et al, Nat Comm, 2019). Here, drawing on a large cohort of 768 MM patients enrolled in the MMRF CoMMpass study (NCT01454297), we comprehensively characterized the landscape of SVs and their functional implications.
Low coverage long-insert WGS (median 4-8X) was available from all patients, of whom 591 also had RNAseq data. Overall, we identified a median of 15 total SVs (range 1-253). Fifty-one percent of SVs (n = 8766) were defined as part of complex events, with a median of one per patient (range 0-14). Chromothripsis, chromoplexy and templated insertions involving >2 chromosomes were observed in 21%, 11% and 21 %, respectively. Chromothripsis was the only SV class with clear prognostic implications after adjustment for molecular and clinical features, resulting in adverse PFS (adjusted HR = 1.57; 95% CI 1.13-2.22; p = 0.008) and OS (adjusted HR = 2.4; 95% CI 1.5-3.83; p < 0.001).
Templated insertions emerged as the cause of CCND1-IGH and MYC translocations in 34 % and 73 % of cases, respectively. This is particularly important given the capability of templated insertions to connect and amplify multiple regions of the genome, involving several oncogenes and regulatory regions (e.g. super enhancers). Twenty-four patients (3.1 %) had translocation between an immunoglobulin locus and a non-canonical driver gene (e.g. PAX5, CD40 and MAP3K14), showing outlier expression by RNAseq where available.
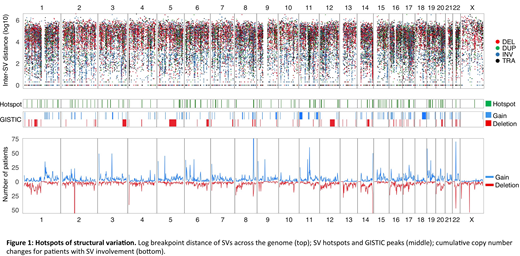
SV hotspot analysis was carried out using the Piecewise Constant Fitting algorithm, comparing the local SV breakpoint density to an empirical background model (Glodzik et al, Nat Genet, 2017). To identify functionally important hotspots, we integrated: 1) local cumulative copy number data, 2) amplification and deletion peaks identified by GISTIC v2 (q < 0.1), 3) gene fusion data and 4) differential expression analysis with adjustment for main molecular subgroups (limma; Bonferroni-Holm adjusted p-values < 0.01). Ninety-eight hotspots were identified (Figure 1), of which 71 (72%) have not previously been reported. Among these novel hotspots, 23 (33 %) contained a known or suspected driver gene, including TNFRSF17 (encoding CAR-T target BCMA), SYK (BCR signal transduction) and KLF2 (key myeloma transcription factor and germline predisposition locus). Active enhancer regions were present in 29 of the novel hotspots (41 %), including 65 % of those with a concurrent putative driver gene involved. For 34 hotspots (48 %) no clear target gene or regulatory region emerged.
SV hotspots and GISTIC peaks covered 13 % of the genome. Overall 38 % of simple and complex SVs had at least one breakpoint falling within a recurrently involved region. The majority of chromoplexy, chromothripsis and templated insertions involved recurrent regions (64, 76 and 86 %, respectively). Simple events were most commonly rare, ranging from 74 % of deletions to 45 % for translocations.
Quantifying the global functional impact of the remaining 72 % of non-recurrent or rare SVs, we observed that genes involved by a rare SV were significantly enriched for outlier expression (z-score +/- 2) compared to a permutation background model. Rare deletions and duplications exerted their effects within 10 Kb of the gene body. Translocations and templated insertions were associated with overexpression up to 1 Mb from the gene, but had no effect when involving the gene body, consistent with a major enhancer hijacking mechanism.
Finally, we sought to understand the role of recurrent and rare SVs in evolutionary dynamics, analyzing 27 patients that progressed with branching evolution. Seventy-two acquired SVs involved a hotspot region (42 driver and/or enhancer; 48 unknown), while 328 were rare.
In conclusion, the SV landscape in multiple myeloma is characterized by multiple recurrently involved genes and regulatory regions. These regions account for the majority of complex SVs, indicating strong positive selection of these events. Nonetheless, the majority of SVs remain unaccounted for. Rare SVs were associated with outlier gene expression and may contribute to the tumor evolutionary trajectory of individual patients.
Papaemmanuil:Celgene: Research Funding. Landgren:Karyopharm: Membership on an entity's Board of Directors or advisory committees; Amgen: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Janssen: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Merck: Other: IDMC; Takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Sanofi: Membership on an entity's Board of Directors or advisory committees; Theradex: Other: IDMC; Adaptive: Honoraria, Membership on an entity's Board of Directors or advisory committees; Abbvie: Membership on an entity's Board of Directors or advisory committees.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal