

TO THE EDITOR:
Venetoclax is an oral, highly selective BCL2 inhibitor approved as monotherapy or in combination with rituximab or obinutuzumab in chronic lymphocytic leukemia (CLL).1-4 Despite complete response rates of up to 50%,1,4,5 secondary resistance is the most frequent cause of treatment failure.6 Resistance mechanisms that have been observed to date in CLL patients treated with venetoclax include (1) early outgrowth of clones with complex karyotype, mutations in BTG1, and aberrations of CDKN2A/B7,8 ; (2) the acquisition of a BCL2 mutation (Gly101Val) that reduces venetoclax binding to BCL29,10 ; and (3) overexpression of other pro-survival proteins BCL-XL and MCL1.9,11 An important observation in all of these scenarios is the subclonality of the mutation-bearing cells within the resistant CLL tumor compartment.9,11 Indeed, the proportion of both the BCL2 Gly101Val resistance mutation and MCL1-overexpressing CLL cells have been reported to vary widely from a minor subclone through to the majority of the tumor compartment.9,11 Moreover, different venetoclax resistance mechanisms (including BCL-XL overexpression and BCL2 Gly101Val mutations) have been observed in independent CLL subpopulations within the same patient.9 Given the observed subclonality of the BCL2 Gly101Val mutation in patients to date and therefore the possibility of additional resistance mechanisms occurring specifically in this subgroup (including a recently described candidate BCL2 resistance mutation Asp103Tyr10 ), we investigated patients with progressive CLL on venetoclax harboring subclonal BCL2 Gly101Val mutations for the presence of additional acquired BCL2 resistance mutations to further explain the clinical resistance of the disease in these patients.
Eleven patients with progressive CLL with BCL2 Gly101Val mutations were identified by sensitive allele-specific droplet digital polymerase chain reaction (ddPCR)9 from among a cohort of 67 patients with heavily pretreated relapsed CLL treated with venetoclax on 3 early-phase clinical trials at our institutions.8 Seven of these patients were described in the original report of BCL2 Gly101Val mutations9 ; 4 patients had newly detected Gly101Val mutations in disease progression samples subsequently (supplemental Material, available on the Blood Web site). Using sample tumor burden assessed by flow cytometry and variant allele frequency (VAF) quantitation determined by ddPCR, the proportion of the CLL tumor compartment harboring BCL2 Gly101Val mutations ranged from a very minor subclone (0.1%) through to the majority of the CLL compartment (68.4%), consistent with previous observations.9,12 The median time from venetoclax commencement to CLL progression in these 11 patients was 36 (range, 13-70) months (additional characteristics are listed in supplemental Table 1).
The BCL2 gene has a high percentage of GC nucleotides resulting in significant technical challenges in variant detection. Therefore we used both (1) digital next-generation sequencing (NGS) using single primer extension and unique molecular indexes to avoid amplicon primer cross-dimerization and perform sequence error correction and (2) hybridization-based target enrichment of BCL2 combined with variant calling by a sensitive tumor-only bioinformatic pipeline optimized for low-level variant calling (supplemental Methods). The estimated limit of detection across the entire BCL2 coding region using this approach was 0.5% VAF representing an approximately 10-fold greater sensitivity and specificity than previous NGS techniques used9 (supplemental Methods).
BCL2 mutations in addition to the Gly101Val were detected in 10 of the 11 patients (91%). A median of 3 mutations (range, 1-7) were observed per patient. Recurrent mutations were observed at the Asp103 codon in 6 patients with amino acid substitutions observed to tyrosine (Tyr), glutamic acid (Glu), and valine (Val) residues (Figure 1A). The Asp103 residue in the P4 pocket is important for hydrogen binding of the azaindole moiety of venetoclax to BCL2 (Figure 1B).13 Other mutations observed in our cohort were Val156Asp (situated at the base of the P2 pocket close to the chlorophenyl moiety of venetoclax) as well as an in-frame insertion (Arg107_Arg110dup) predicted to duplicate and lengthen the intervening 4 amino acid sequence that separates the α2 and α3 helices (Figure 1B). The Asp103/Val156 substitutions and the Arg107_Arg110dup have not previously been described in cancer databases (COSMIC, https://cancer.sanger.ac.uk/cosmic) or the literature, to our knowledge, outside the single previous case report of the Asp103Tyr occurring in a patient with CLL treated with venetoclax.10 As with the Gly101Val, these observations support the specificity of these mutations for the context of venetoclax resistance. In addition, Ala113Gly and Arg129Leu mutations were observed. Both of these mutations have previously been observed in B-cell lymphomas,14,15 with the Arg129 being a recurrently mutated codon in BCL2 in lymphoid malignancy (COSMIC). Importantly, these mutations were not observed in a cohort of 96 venetoclax-naïve patients with CLL.9 Asp103Glu/Tyr codon variants, Val156Asp and Arg107_Arg110dup, were orthogonally validated using allele-specific ddPCR assays and were not detectable in available samples from 6 patients collected before exposure to venetoclax (supplemental Methods).
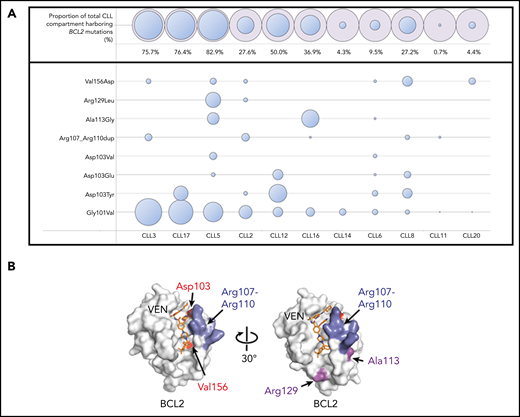
BCL2 mutations in patients with progressive CLL on venetoclax. (A) BCL2 mutations in a cohort of patients with CLL progression on venetoclax. Patients are ordered in descending Gly101Val cancer cell fraction (CCF). CCF was determined as (VAF/disease burden determined by flow cytometry) × 2 (assuming heterozygosity). Area of blue circles is proportional to CCF mutated. The top row shows the total CCF harboring BCL2 mutations (the sum of individual CCF and assumes occurrence in mutually exclusive cells). Mutations detected were c.302G>T, p.(Gly101Val); c.302_303delinsTT, p.(Gly101Val); c.307G>T, p.(Asp103Tyr); c.308A>T, p.(Asp103Val); c.309C>A, p.(Asp103Glu); c.319_330dup, p.(Arg107_Arg110dup); c.338C>G, p.(Ala113Gly); c.386G>T, p.(Arg129Leu); c.467T>A, p.(Val156Asp); BCL2 NM_000633.2. Patient CLL6 had both a c.302G>T and a complex variant (c.302_303delinsTT) leading to a p.(Gly101Val) in different reads; the Gly101Val area is the sum of the 2 CCFs for this patient. (B) Structure of BCL2 protein with venetoclax bound (PDB ID 6O0K) illustrating the positions of the mutated residues Asp103, Val156, Arg107 to Arg110, Ala113, and Arg129.
BCL2 mutations in patients with progressive CLL on venetoclax. (A) BCL2 mutations in a cohort of patients with CLL progression on venetoclax. Patients are ordered in descending Gly101Val cancer cell fraction (CCF). CCF was determined as (VAF/disease burden determined by flow cytometry) × 2 (assuming heterozygosity). Area of blue circles is proportional to CCF mutated. The top row shows the total CCF harboring BCL2 mutations (the sum of individual CCF and assumes occurrence in mutually exclusive cells). Mutations detected were c.302G>T, p.(Gly101Val); c.302_303delinsTT, p.(Gly101Val); c.307G>T, p.(Asp103Tyr); c.308A>T, p.(Asp103Val); c.309C>A, p.(Asp103Glu); c.319_330dup, p.(Arg107_Arg110dup); c.338C>G, p.(Ala113Gly); c.386G>T, p.(Arg129Leu); c.467T>A, p.(Val156Asp); BCL2 NM_000633.2. Patient CLL6 had both a c.302G>T and a complex variant (c.302_303delinsTT) leading to a p.(Gly101Val) in different reads; the Gly101Val area is the sum of the 2 CCFs for this patient. (B) Structure of BCL2 protein with venetoclax bound (PDB ID 6O0K) illustrating the positions of the mutated residues Asp103, Val156, Arg107 to Arg110, Ala113, and Arg129.
In the 10 patients with multiple BCL2 mutations, all mutations were observed to be present in different reads in NGS data, consistent with their presence in different cells (assuming heterozygosity). To exclude the presence of BCL2 locus amplification accounting for multiple mutations in the same cell, we performed copy number analysis using read abundance from hybridization sequencing, as described previously,16 and did not detect BCL2 copy number gains in any of these patients. Interestingly, a focal copy number gain at the MCL1 locus was detected in patient CLL16, which was not detectable in the pre-venetoclax sample (supplemental Figure 1). Moreover, patient CLL2 has previously had BCL-XL overexpression in cells not containing the Gly101Val mutation.9 Therefore, across this cohort, we have observed both a range of BCL2 mutations (accounting for an estimated total cancer cell fraction ranging from 0.7% to 82.9%) as well as less frequent concomitant aberrations leading to overexpression of alternative pro-survival proteins (Figure 1A).
The BCL2 Asp103Glu mutation, observed in 4 patients, is particularly noteworthy. The equivalent residue to BCL2 Asp103 is Glu in BCL-XL and is one of the crucial determinants for the selectivity of venetoclax for BCL2 over BCL-XL.13 Thus, Asp103Glu is predicted to alter the P4-binding pocket to more closely resemble that of BCL-XL. Given this, we hypothesized that the Asp103Glu mutation may impart differential sensitivity to venetoclax, which selectively targets BCL2, and to navitoclax, which also targets BCL-XL. We first investigated the in vitro binding profile of the Asp103Glu to both venetoclax and pro‐apoptotic proteins in competition surface plasmon resonance binding experiments. BCL2 Asp103Glu was associated with a decrease in affinity for venetoclax of approximately 20-fold compared with the wild-type aspartic acid (Gly101Val showed an ∼180-fold reduction9 ; supplemental Table 2). No reduction was observed for binding to a BIM BH3-only peptide, consistent with previous observations and that BCL2 resistance mutations are selected on the ability to maintain cell survival by binding and sequestering BH3-only proteins (eg, BIM), while selectively reducing affinity for venetoclax17 (Figure 2A). We then went on to express the Asp103Glu in 2 B-lineage human cell lines: RS4;11 (acute leukemia) and KMS-12-PE (myeloma). As with Gly101Val, Asp103Glu-expressing cells were markedly less sensitive to venetoclax (Figure 2B). In sharp contrast, Asp103Glu-expressing cells displayed heightened sensitivity to navitoclax consistent with Asp103 conferring venetoclax selectivity for BCL2 (Figure 2B).18
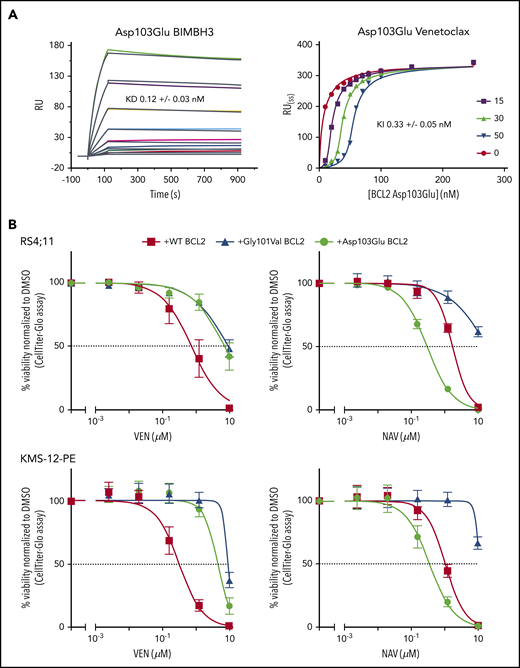
Venetoclax/BIM binding characteristics and in vitro sensitivity of BCL2 Asp103Glu mutation. (A) Impact of Asp103Glu on the ability of BCL2 to bind BH3 ligands. (Left) BIMBH3 binding. A total of 0 to 40 nmol/L mutant BCL2 was used as an analyte against the BIMBH3 peptide immobilized on a surface plasmon resonance (BIAcore) sensor chip. The raw response (RU) curves (colored curves) from a representative experiment were fitted to 1 site-specific kinetic model (black curves) to derive on and off rates (supplemental Table 2), and calculate KD values for interactions with Asp103Glu. (Right) Steady-state competition of various venetoclax concentrations (0-50 nmol/L) prebound to Asp103Glu (0-250 nmol/L), competing against a BIMBH3 immobilized chip. Fitted data were used to derive the KI for venetoclax binding to Asp103Glu. Data are representative of 3 independent experiments, reporting means ± 1 SD. (B) Expression of BCL2 Asp103Glu in RS4;11 (top) or KMS-12-PE (bottom) cell lines reduces sensitivity to venetoclax (left) but not to navitoclax (right). Each of these mutants or wild-type (WT) BCL2 were expressed and the in vitro sensitivities to venetoclax (0-10 μmol/L) or to navitoclax (NAV; 0-10 μmol/L) measured 24 hours later. Data represent means ± 1 SD of at least 3 independent experiments. KD,equilibrium binding constant for BIMBH3 peptide, from direct binding experiments; KI, fitted equilibrium binding constant for venetoclax, from steady-state competition experiments.
Venetoclax/BIM binding characteristics and in vitro sensitivity of BCL2 Asp103Glu mutation. (A) Impact of Asp103Glu on the ability of BCL2 to bind BH3 ligands. (Left) BIMBH3 binding. A total of 0 to 40 nmol/L mutant BCL2 was used as an analyte against the BIMBH3 peptide immobilized on a surface plasmon resonance (BIAcore) sensor chip. The raw response (RU) curves (colored curves) from a representative experiment were fitted to 1 site-specific kinetic model (black curves) to derive on and off rates (supplemental Table 2), and calculate KD values for interactions with Asp103Glu. (Right) Steady-state competition of various venetoclax concentrations (0-50 nmol/L) prebound to Asp103Glu (0-250 nmol/L), competing against a BIMBH3 immobilized chip. Fitted data were used to derive the KI for venetoclax binding to Asp103Glu. Data are representative of 3 independent experiments, reporting means ± 1 SD. (B) Expression of BCL2 Asp103Glu in RS4;11 (top) or KMS-12-PE (bottom) cell lines reduces sensitivity to venetoclax (left) but not to navitoclax (right). Each of these mutants or wild-type (WT) BCL2 were expressed and the in vitro sensitivities to venetoclax (0-10 μmol/L) or to navitoclax (NAV; 0-10 μmol/L) measured 24 hours later. Data represent means ± 1 SD of at least 3 independent experiments. KD,equilibrium binding constant for BIMBH3 peptide, from direct binding experiments; KI, fitted equilibrium binding constant for venetoclax, from steady-state competition experiments.
Although further comprehensive genomic assessment beyond BCL2 in this cohort would be required to fully understand all contributions to resistance, the detection of multiple BCL2 mutations per patient suggests that alternative approaches to persisting with long-term venetoclax may be preferable. The use of initial time-limited exposure is now an established practice and is effective particularly in those who achieve negative measurable residual disease (MRD).4,5 However, for patients with persisting MRD after initial therapy and those in which a BCL2 mutation is detected in the presence of rising MRD, the addition of other agents will be required (potentially including alternative BH3-mimetic agents such as navitoclax).
In summary, we have identified multiple novel BCL2 mutations acquired in parallel with BCL2 Gly101Val during venetoclax therapy. Collectively, these BCL2 mutations account for a greater proportion of the resistant compartment in individual patients than previously recognized. Our observations and experimental data are consistent with the Asp103 codon variants imparting resistance to venetoclax, but not necessarily navitoclax in vitro. Finally, our observation of the presence of multiple BCL2 mutations, as well as simultaneous aberrations in BCL-XL and MCL1, further consolidates the paradigm emerging across hematological malignancies19 of multiple, independent molecular mechanisms underpinning an “oligoclonal” pattern of clinical relapse on targeted therapies.
For original data, please e-mail the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors are grateful to the patients who enrolled on the venetoclax clinical trials, and for assistance from Naomi Sprigg with collection and curation of samples.
This research was supported by grants from the Snowdome Foundation (P.B. and M.A.A.), Vision Super and the Wilson Centre for Lymphoma Genomics (P.B. and D.A.W.), the Leukemia and Lymphoma Society (fellowship 5467-18 [R.T.], SCOR 7015-18 [D.C.S.H. and A.W.R.]), the National Health and Medical Research Council of Australia (fellowships 1079700 [P.E.C.], 1043149 [D.C.S.H], and 1079560 [A.W.R.]), and grants to project 1141874 (P.E.C.), program 1113133 (D.C.S.H.), and program 1113577 (A.W.R.). This work was made possible through Victorian State Government Operational Infrastructure Support and Australian Government National Health and Medical Research Council Independent Research Institute Infrastructure Support Scheme.
Authorship
Contribution: P.B. designed research, performed research, analyzed data, and wrote the paper; E.R.T. and R.W.B. analyzed data, performed research, and wrote the paper; T.N., J.G., X.C., M.M., and R.T. performed research and analyzed data; T.C. contributed bioinformatics tools; M.A.A. and J.F.S. provided clinical care to patients and wrote the paper; D.A.W. and P.E.C. analyzed data and wrote the paper; D.C.S.H. designed research, analyzed data, and wrote the paper; A.W.R. provided clinical care for patients, designed research, and wrote the paper; and all authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: M.A.A., J.G., R.T., R.W.B., P.E.C., D.C.S.H., and A.W.R. are employees of the Walter and Eliza Hall Institute, which receives milestone and royalty payments related to venetoclax; M.A.A., J.G., P.E.C., D.C.S.H., and A.W.R. receive financial benefits related to these payments. J.F.S. has received research funding from AbbVie and Genentech and is a consultant and member of advisory boards for both companies. D.C.S.H. has received research funding from Genentech. A.W.R. has received research funding from AbbVie. The remaining authors declare no competing financial interests.
Correspondence: Piers Blombery, Department of Pathology, Peter MacCallum Cancer Centre, 305 Grattan St, Melbourne, VIC 3000, Australia; e-mail: piers.blombery@petermac.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal