

Key Points
The ratio of plasma A1M to hemopexin concentration is associated with AKI biomarkers in SCD.
Enhanced scavenging of circulating heme to kidney triggers AKI in transgenic SCD mice.

Abstract
Acute kidney injury (AKI) is a major clinical concern in sickle cell disease (SCD). Clinical evidence suggests that red cell alarmins may cause AKI in SCD, however, the sterile inflammatory process involved has hitherto not been defined. We discovered that hemopexin deficiency in SCD is associated with a compensatory increase in α-1-microglobulin (A1M), resulting in an up to 10-fold higher A1M-to-hemopexin ratio in SCD compared with healthy controls. The A1M-to-hemopexin ratio is associated with markers of hemolysis and AKI in both humans and mice with SCD. Studies in mice showed that excess heme is directed to the kidneys in SCD in a process involving A1M causing AKI, whereas excess heme in controls is transported to the liver as expected. Using genetic and bone marrow chimeric tools, we confirmed that hemopexin deficiency promotes AKI in sickle mice under hemolytic stress. However, AKI was blocked when hemopexin deficiency in sickle mice was corrected with infusions of purified hemopexin prior to the induction of hemolytic stress. This study identifies acquired hemopexin deficiency as a risk factor of AKI in SCD and hemopexin replacement as a potential therapy.
Introduction
Acute kidney injury (AKI) is a common complication during vaso-occlusive crisis (VOC) and acute chest syndrome (ACS) in patients with sickle cell disease (SCD).1-3 A major clinical concern with AKI is that it is a risk factor for chronic kidney disease and end-stage renal disease, which together account for 16% to 18% of SCD mortality.4,5 A rapid decline in hemoglobin (Hb) is associated with AKI in SCD,2 suggesting that red cell alarmins released during hemolysis may cause kidney injury.6,7 Among this group of alarmins, extracellular heme has been shown to induce ACS and VOC in humanized murine models of SCD,8,9 suggesting that there may be a common sterile inflammatory pathway involving heme that links VOC and ACS to AKI.
Hemopexin and α-1-microglobulin (A1M) are the major plasma proteins that scavenge extracellular heme from the blood circulation. Heme is transported to the liver by hemopexin and degraded by heme oxygenases.10 There is an acquired deficiency of hemopexin in SCD due to the intensity and chronicity of hemolysis.11 Thus, SCD patients may be vulnerable to heme-mediated tissue injury during acute hemolysis. The second heme scavenger A1M is a 26-kDa protein. Heme-bound A1M passes through the glomerular filtration system because of its small size, indicating that renal tubular epithelium becomes exposed to heme during its removal from circulation.12,13 We reasoned that a disproportionately larger amount of heme may be cleared through the kidneys in SCD during acute hemolytic stress because of the acquired hemopexin deficiency. We tested this hypothesis studying patients at steady state, and a clinically relevant transgenic mouse model of SCD. Our results reveal a novel insight into the pathogenesis of AKI in SCD, and we provide proof of principle in how to prevent this complication in a mouse model.
Study design
Reagents
Hemin [Fe(III)PPIX; Sigma-Aldrich] was prepared as reported.8 Purified human hemopexin (Athens Research) and human A1M (Bio-Rad) were obtained commercially.
Patient samples
Plasma, serum, and urinary samples were obtained from patients at steady state in the ORDIS Study of the Sickle Cell Disease Genomics Network of Africa (SickleGenAfrica Network), approved by ethics committees of the Kwame Nkrumah University of Science and Technology, Ghana (CHRPE/AP/325/14, CHRPE/AP/104/16, and CHRPE/140/17) and the University of Pittsburgh Institutional Review Board (PRO14010452).14 Research was conducted in accordance with the Declaration of Helsinki.
Mice
The following mice were purchased from The Jackson Laboratory and studied at the University of Pittsburgh (Institutional Animal Care and Use Committee approval #18072763): knockin Townes expressing human Hb βS (SS) and human Hb βA (AA),15 hemopexin-null (HxKO), and C57BL/6 (HxWT). Hemopexin-null and C57BL/6 mice were transplanted with SS bone marrow as we have reported previously.16 Total Hb was measured using the AVOXImeter (ITCmed) and reticulocytes by flow cytometry.
Biochemical analysis
Plasma Hb, lactate dehydrogenase (LDH), and tissue heme content were measured using assays from Bioassay Systems. Creatinine (Cr) and albumin were measured using assays from Arbor Assays. Enzyme-linked immunosorbent assay (ELISA) was used to measure levels of hemopexin (Kamiya Biomedical), A1M (BioLegend), kidney injury molecule-1 (KIM-1), and neutrophil gelatinase–associated lipocalin (NGAL) (R&D Systems).
Glomerular filtration rate
A transcutaneous device (MediBeacon) was used to monitor clearance of fluorescein isothiocyanate (FITC)-sinistrin infused into mice (7.5 mg/100 g) and the data were plotted relative to the percentage of maximum fluorescence to measure glomerular filtration rate (GFR).17
Histopathology
Kidney tissue sections were stained with hematoxylin and eosin, periodic acid–Schiff, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (Trevigen) as well as Perls Prussian Blue and examined using an Olympus AX70 microscope; histopathological scoring was performed as previously described.18,19
Statistical analysis
The 2-tailed, paired or unpaired Student t test, and 2-way analysis of variance were performed. GraphPad Prism 7 software was used.
Results and discussion
Low hemopexin level is associated with increased A1M in SCD
Although hemopexin deficiency is a well-established paradigm in SCD, the level of A1M, the second heme scavenger, has never been reported. We found that A1M is markedly elevated among SCD patients; there was a 4.4-fold reduction in hemopexin and a 1.6-fold increase of plasma A1M in a cohort of SCD patients compared with healthy controls (Figure 1A-B). Because A1M transports heme to the kidneys, we explored whether raised A1M is associated with kidney injury in SCD patients. The molar concentration of A1M and hemopexin (A1M/hemopexin) correlated with hemolytic markers, plasma Hb and serum LDH, in a cohort of SCD patients at steady state14 (Figure 1C-D). The A1M-to-hemopexin ratio correlated with urinary KIM-1 and serum NGAL, 2 AKI biomarkers associated with hemolysis and known to be elevated in SCD patients20 (Figure 1E-F). These results indicate a compensatory increase in A1M in SCD that is associated with kidney injury markers.
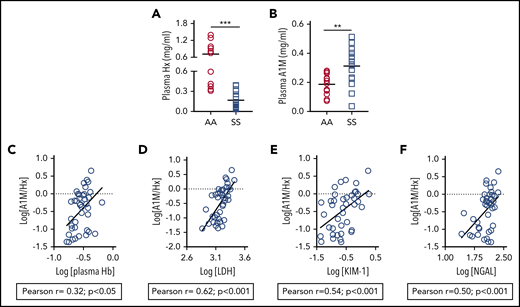
Plasma A1M is raised in association with hemolytic and AKI biomarkers in SCD patients. (A) Level of plasma A1M is elevated in patients with SCD (SS) compared with age- and ethnically matched Hb AA individuals (n = 12-15). (B) Level of plasma hemopexin (Hx) in the same cohort of plasma as in panel A. (C-F) Whole-blood samples and urine were collected at the same time from SCD patients (n = 40) at steady state. Blood samples were processed to collect serum and plasma. Plasma A1M and hemopexin, plasma Hb, serum LDH activity, urinary KIM-1, and serum NGAL were measured using specific colorimetric or ELISA kits following the manufacturer’s instructions. Association of molar concentration of A1M and hemopexin (A1M/Hx) with (C) plasma Hb, (D) serum LDH, (E) urinary KIM-1, and (F) serum NGAL in logarithmic scale.
Plasma A1M is raised in association with hemolytic and AKI biomarkers in SCD patients. (A) Level of plasma A1M is elevated in patients with SCD (SS) compared with age- and ethnically matched Hb AA individuals (n = 12-15). (B) Level of plasma hemopexin (Hx) in the same cohort of plasma as in panel A. (C-F) Whole-blood samples and urine were collected at the same time from SCD patients (n = 40) at steady state. Blood samples were processed to collect serum and plasma. Plasma A1M and hemopexin, plasma Hb, serum LDH activity, urinary KIM-1, and serum NGAL were measured using specific colorimetric or ELISA kits following the manufacturer’s instructions. Association of molar concentration of A1M and hemopexin (A1M/Hx) with (C) plasma Hb, (D) serum LDH, (E) urinary KIM-1, and (F) serum NGAL in logarithmic scale.
Maladaptive removal of excess heme from the blood triggers AKI in sickle mice
Transgenic sickle mice phenocopied the hemopexin/A1M dynamics in humans; the A1M-to-hemopexin ratio was sevenfold higher in SS mice compared with AA controls (Figure 2A). To test the clinical implications of this finding, we infused a low concentration of purified heme (hemin; 20 μmol/kg) into the mice, and discovered that the excess heme was delivered to the liver in the control animals. By contrast, the excess heme in the SS mice was deposited to the kidneys and not the liver (Figure 2B). The plasma Cr and urinary albumin-Cr ratio (uACR) increased fourfold and twofold, respectively, compared with baseline levels in the SS mice after the heme challenge, whereas no change occurred in the control mice (Figure 2C-D). AKI in the SS mice was supported by elevations in urinary KIM-1, and in plasma and urinary NGAL (Figure 2E; supplemental Figure 1A-B, available on the Blood Web site). Because AKI is defined functionally by a rapid decline in GFR,21 we assessed GFR in the mice by measuring renal clearance of FITC-sinistrin. These studies revealed an ∼28% reduction in GFR following a challenge with hemin compared with baseline in the SS and no change in the control AA mice (Figure 2F-H; supplemental Figure 1C). Histopathological analysis revealed severe damage in the renal cortex and medulla, and tubular cell death exclusively in the SS mice (Figure 2I-L; supplemental Figure 1E). Increased iron in the renal tubular cells suggests that iron may have contributed to the cell death in the SS mouse kidneys (supplemental Figure 1F). These results indicate that a modest increase in circulating heme that has no effect in healthy animals can cause AKI in SCD.
![Disproportionate transport of excess circulating heme to kidney induces AKI in sickle mice. (A) Plasma concentration of A1M is elevated in sickle transgenic (SS) mice compared with control (AA) mice (n = 8-12). Ratio of molar concentration of A1M and hemopexin (A1M/Hx) in plasma from SS and AA mice (n = 8-12). (B-L) Transgenic sickle (SS) and littermate control (AA) mice were infused with hemin (20 μmol/kg body weight [BW]). (B) Total heme content in kidney and liver tissue 2 days following infusion of hemin (H) or vehicle (V) in sickle transgenic mice (SS) and control (AA) mice (n = 6). Heme was scavenged preferentially to the kidneys in SS mice, and to the liver in AA mice. (C) Paired analysis of plasma Cr in AA and SS mice (n = 6) at baseline (BL) and 2 days (D2) following infusion of exogenous hemin. (D) Ratio of albumin and Cr in spot urine (uACR) collected from the same cohort of mice as in panel C. (E) ELISA of urine samples from AA and SS mice, showing an ∼3.6-fold increase in urinary KIM-1 in SS mice following hemin infusion (n = 8-12). (F-G) Relative fluorescence showing real-time clearance of infused FITC-sinstrin from circulation in (F) AA mice and (G) SS mice prior to and 2 days following hemin infusion. Delayed clearance of FITC-sinistrin in hemin-infused SS mice indicates reduced renal function. (H) GFR representing kidney function in AA and SS mice at baseline and following hemin infusion (n = 4-7). (I) Representative images of cortex and medulla from hematoxylin-and-eosin–stained renal sections of AA and SS mice challenged with vehicle or hemin showing cortical tubular vacuolation and atrophy with severe medullary peritubular capillary congestion in heme-infused SS mice kidney (original magnification ×400). (J) Semiquantitative histological kidney injury score in AA and SS mice in panel I. (K) Represented renal histology following TUNEL staining (blue stain indicates fragmented DNA content) showing tubular cell death/apoptosis in SS mice kidney tubules (original magnification ×400). (L) Semiquantitative apoptotic index showing percentage of TUNEL+ tubules in panelK. (M) Control (HxWT) mice and mice lacking hemopexin expression (HxKO) were transplanted with bone marrow from SS mice to create sickle mice with (HxWTSS) or without (HxKOSS) hemopexin in nonhematopoietic tissues. Level of plasma hemopexin (Hx; left y-axis) and plasma A1M (right y-axis) in HxWT, HxKO, HxWTSS, and HxKOSS mice showing that 2 heme scavengers are inversely related (n = 3-5). (N) Paired analysis of GFR at baseline and following hemin (20 μmol/kg BW) challenge (H) in indicated group of mice (n = 3-5) showing that absence of plasma hemopexin severely impairs renal function in SS mice leading to AKI. (O) SS mice were treated with hemopexin (1 mg per mouse) or A1M (0.25 mg per mouse) or vehicle immediately prior to hemin (20 μmol/kg BW) infusion. Calculated GFR at baseline and 2 days following hemin infusion in SS mice treated with vehicle, hemopexin, or A1M showing protection from AKI development in hemopexin-treated mice. *P < .05, **P < .01, ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/13/10.1182_blood.2019002653/4/m_bloodbld2019002653f2-1.png?Expires=1767710460&Signature=eC2VTcvVu7sE4BnJ5NR4IqbLK9VVxKKdxupVYWfRbSpeX3YtP6~MfwhWDJbV-GuR7l6jrEkeu~eSuH2GqSjI4yydBJ63Hl7w2I9NFdQ8AfRh2lSZgD~RH4oMT4Q9pkRezveElM2xeH0JpVN1fkY8Cj2wEhbNPFX~JaK2bO9iT4n~~~nTfK0POVCi3RTLwa-vOCySUgKc99ok94zEJx2bnWKagXPsP0-u~sAC5I8wqFA8JBMixLJXKXG0jzyY3xV6bSzWRkYJQchBoStDx9uiE-dPEPbFJ~Z~zDKZLdiBmYpklnzfjMmdimoUQS10HSNbnH9PMPdY-vkUPVeCAFjVTQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![Disproportionate transport of excess circulating heme to kidney induces AKI in sickle mice. (A) Plasma concentration of A1M is elevated in sickle transgenic (SS) mice compared with control (AA) mice (n = 8-12). Ratio of molar concentration of A1M and hemopexin (A1M/Hx) in plasma from SS and AA mice (n = 8-12). (B-L) Transgenic sickle (SS) and littermate control (AA) mice were infused with hemin (20 μmol/kg body weight [BW]). (B) Total heme content in kidney and liver tissue 2 days following infusion of hemin (H) or vehicle (V) in sickle transgenic mice (SS) and control (AA) mice (n = 6). Heme was scavenged preferentially to the kidneys in SS mice, and to the liver in AA mice. (C) Paired analysis of plasma Cr in AA and SS mice (n = 6) at baseline (BL) and 2 days (D2) following infusion of exogenous hemin. (D) Ratio of albumin and Cr in spot urine (uACR) collected from the same cohort of mice as in panel C. (E) ELISA of urine samples from AA and SS mice, showing an ∼3.6-fold increase in urinary KIM-1 in SS mice following hemin infusion (n = 8-12). (F-G) Relative fluorescence showing real-time clearance of infused FITC-sinstrin from circulation in (F) AA mice and (G) SS mice prior to and 2 days following hemin infusion. Delayed clearance of FITC-sinistrin in hemin-infused SS mice indicates reduced renal function. (H) GFR representing kidney function in AA and SS mice at baseline and following hemin infusion (n = 4-7). (I) Representative images of cortex and medulla from hematoxylin-and-eosin–stained renal sections of AA and SS mice challenged with vehicle or hemin showing cortical tubular vacuolation and atrophy with severe medullary peritubular capillary congestion in heme-infused SS mice kidney (original magnification ×400). (J) Semiquantitative histological kidney injury score in AA and SS mice in panel I. (K) Represented renal histology following TUNEL staining (blue stain indicates fragmented DNA content) showing tubular cell death/apoptosis in SS mice kidney tubules (original magnification ×400). (L) Semiquantitative apoptotic index showing percentage of TUNEL+ tubules in panelK. (M) Control (HxWT) mice and mice lacking hemopexin expression (HxKO) were transplanted with bone marrow from SS mice to create sickle mice with (HxWTSS) or without (HxKOSS) hemopexin in nonhematopoietic tissues. Level of plasma hemopexin (Hx; left y-axis) and plasma A1M (right y-axis) in HxWT, HxKO, HxWTSS, and HxKOSS mice showing that 2 heme scavengers are inversely related (n = 3-5). (N) Paired analysis of GFR at baseline and following hemin (20 μmol/kg BW) challenge (H) in indicated group of mice (n = 3-5) showing that absence of plasma hemopexin severely impairs renal function in SS mice leading to AKI. (O) SS mice were treated with hemopexin (1 mg per mouse) or A1M (0.25 mg per mouse) or vehicle immediately prior to hemin (20 μmol/kg BW) infusion. Calculated GFR at baseline and 2 days following hemin infusion in SS mice treated with vehicle, hemopexin, or A1M showing protection from AKI development in hemopexin-treated mice. *P < .05, **P < .01, ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/13/10.1182_blood.2019002653/4/m_bloodbld2019002653f2-2.png?Expires=1767710460&Signature=yLWjZGVjhrY9CdPOINCcVSkTWIUfW-Lxqc8JF3K0ySsI8j-RLLvOnX5XCsCk~P7YCHRLV3km75pA05lxvBXbShO2smh2opy8MVrYVw3O1dQYnENafSGrYVh0jJ0LLcFL14dSLV4DGkdbabgRdYVB1BCeEIn9nF2ozy6q5lu~TWnjWEmdy49ItTAHcsXFoapVxEoCxGnb7cEEuU9PPXk2w4f3q6-QkmG~YMbr0H9JcUiRohduxUHU~bpcfq40QtzqmjVeA07eabQXUZNaWsNUSnVQiC8QmtjmzNWOPu-YepIXEtyFApcuLMKtvcTzbHWTR8bLBS1uQc~2L4SOCRgrRg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Disproportionate transport of excess circulating heme to kidney induces AKI in sickle mice. (A) Plasma concentration of A1M is elevated in sickle transgenic (SS) mice compared with control (AA) mice (n = 8-12). Ratio of molar concentration of A1M and hemopexin (A1M/Hx) in plasma from SS and AA mice (n = 8-12). (B-L) Transgenic sickle (SS) and littermate control (AA) mice were infused with hemin (20 μmol/kg body weight [BW]). (B) Total heme content in kidney and liver tissue 2 days following infusion of hemin (H) or vehicle (V) in sickle transgenic mice (SS) and control (AA) mice (n = 6). Heme was scavenged preferentially to the kidneys in SS mice, and to the liver in AA mice. (C) Paired analysis of plasma Cr in AA and SS mice (n = 6) at baseline (BL) and 2 days (D2) following infusion of exogenous hemin. (D) Ratio of albumin and Cr in spot urine (uACR) collected from the same cohort of mice as in panel C. (E) ELISA of urine samples from AA and SS mice, showing an ∼3.6-fold increase in urinary KIM-1 in SS mice following hemin infusion (n = 8-12). (F-G) Relative fluorescence showing real-time clearance of infused FITC-sinstrin from circulation in (F) AA mice and (G) SS mice prior to and 2 days following hemin infusion. Delayed clearance of FITC-sinistrin in hemin-infused SS mice indicates reduced renal function. (H) GFR representing kidney function in AA and SS mice at baseline and following hemin infusion (n = 4-7). (I) Representative images of cortex and medulla from hematoxylin-and-eosin–stained renal sections of AA and SS mice challenged with vehicle or hemin showing cortical tubular vacuolation and atrophy with severe medullary peritubular capillary congestion in heme-infused SS mice kidney (original magnification ×400). (J) Semiquantitative histological kidney injury score in AA and SS mice in panel I. (K) Represented renal histology following TUNEL staining (blue stain indicates fragmented DNA content) showing tubular cell death/apoptosis in SS mice kidney tubules (original magnification ×400). (L) Semiquantitative apoptotic index showing percentage of TUNEL+ tubules in panelK. (M) Control (HxWT) mice and mice lacking hemopexin expression (HxKO) were transplanted with bone marrow from SS mice to create sickle mice with (HxWTSS) or without (HxKOSS) hemopexin in nonhematopoietic tissues. Level of plasma hemopexin (Hx; left y-axis) and plasma A1M (right y-axis) in HxWT, HxKO, HxWTSS, and HxKOSS mice showing that 2 heme scavengers are inversely related (n = 3-5). (N) Paired analysis of GFR at baseline and following hemin (20 μmol/kg BW) challenge (H) in indicated group of mice (n = 3-5) showing that absence of plasma hemopexin severely impairs renal function in SS mice leading to AKI. (O) SS mice were treated with hemopexin (1 mg per mouse) or A1M (0.25 mg per mouse) or vehicle immediately prior to hemin (20 μmol/kg BW) infusion. Calculated GFR at baseline and 2 days following hemin infusion in SS mice treated with vehicle, hemopexin, or A1M showing protection from AKI development in hemopexin-treated mice. *P < .05, **P < .01, ***P < .001.
Disproportionate transport of excess circulating heme to kidney induces AKI in sickle mice. (A) Plasma concentration of A1M is elevated in sickle transgenic (SS) mice compared with control (AA) mice (n = 8-12). Ratio of molar concentration of A1M and hemopexin (A1M/Hx) in plasma from SS and AA mice (n = 8-12). (B-L) Transgenic sickle (SS) and littermate control (AA) mice were infused with hemin (20 μmol/kg body weight [BW]). (B) Total heme content in kidney and liver tissue 2 days following infusion of hemin (H) or vehicle (V) in sickle transgenic mice (SS) and control (AA) mice (n = 6). Heme was scavenged preferentially to the kidneys in SS mice, and to the liver in AA mice. (C) Paired analysis of plasma Cr in AA and SS mice (n = 6) at baseline (BL) and 2 days (D2) following infusion of exogenous hemin. (D) Ratio of albumin and Cr in spot urine (uACR) collected from the same cohort of mice as in panel C. (E) ELISA of urine samples from AA and SS mice, showing an ∼3.6-fold increase in urinary KIM-1 in SS mice following hemin infusion (n = 8-12). (F-G) Relative fluorescence showing real-time clearance of infused FITC-sinstrin from circulation in (F) AA mice and (G) SS mice prior to and 2 days following hemin infusion. Delayed clearance of FITC-sinistrin in hemin-infused SS mice indicates reduced renal function. (H) GFR representing kidney function in AA and SS mice at baseline and following hemin infusion (n = 4-7). (I) Representative images of cortex and medulla from hematoxylin-and-eosin–stained renal sections of AA and SS mice challenged with vehicle or hemin showing cortical tubular vacuolation and atrophy with severe medullary peritubular capillary congestion in heme-infused SS mice kidney (original magnification ×400). (J) Semiquantitative histological kidney injury score in AA and SS mice in panel I. (K) Represented renal histology following TUNEL staining (blue stain indicates fragmented DNA content) showing tubular cell death/apoptosis in SS mice kidney tubules (original magnification ×400). (L) Semiquantitative apoptotic index showing percentage of TUNEL+ tubules in panelK. (M) Control (HxWT) mice and mice lacking hemopexin expression (HxKO) were transplanted with bone marrow from SS mice to create sickle mice with (HxWTSS) or without (HxKOSS) hemopexin in nonhematopoietic tissues. Level of plasma hemopexin (Hx; left y-axis) and plasma A1M (right y-axis) in HxWT, HxKO, HxWTSS, and HxKOSS mice showing that 2 heme scavengers are inversely related (n = 3-5). (N) Paired analysis of GFR at baseline and following hemin (20 μmol/kg BW) challenge (H) in indicated group of mice (n = 3-5) showing that absence of plasma hemopexin severely impairs renal function in SS mice leading to AKI. (O) SS mice were treated with hemopexin (1 mg per mouse) or A1M (0.25 mg per mouse) or vehicle immediately prior to hemin (20 μmol/kg BW) infusion. Calculated GFR at baseline and 2 days following hemin infusion in SS mice treated with vehicle, hemopexin, or A1M showing protection from AKI development in hemopexin-treated mice. *P < .05, **P < .01, ***P < .001.
To confirm our findings in the SS mice, we studied a genetic mouse model with no expression of hemopexin (HxKO mice). We confirmed that HxKO have no detectable hemopexin, however, these animals had raised A1M. We created bone marrow chimeric mice with the SCD phenotype on the HxKO and HxWT backgrounds (supplemental Figure 2A-B). As expected, hemopexin levels were low in both SCD chimeras whereas A1M was raised (Figure 2M). The HxKOSS mice had the lowest baseline GFR and increased KIM-1 compared with HxKO and HxWTSS mice. There was no significant difference in the level of uACR in the chimeric or wild-type mice (Figure 2N; supplemental Figure 2C-D). Induction of AKI resulted in a significant reduction in GFR in the HxKO, HxWTSS, and HxKOSS mice, confirming the risk low hemopexin poses to the kidneys during hemolytic stress. Two-way analysis of variance revealed a worse drop in GFR and uACR for the HxKOSS mice compared with the HxKO and HxWTSS mice (Figure 2N). The HxKOSS mice also had significantly increased uACR and threefold renal heme accumulation compared with the HxWTSS mouse following heme infusion (supplemental Figure 2E-F).
To determine whether hemopexin can block AKI in SCD, we infused SS mice with purified human hemopexin, A1M, or vehicle immediately prior to AKI induction. The hemopexin infusion stabilized GFR whereas A1M exacerbated renal impairment by 39%. Hemopexin treatment also blocked changes in plasma Cr, uACR and uKIM-1, and renal tissue injury when SS mice were challenged with hemin (Figure 2O; supplemental Figure 3A-D).
In summary, we have discovered that the acquired hemopexin deficiency in SCD is a risk factor for AKI. The relative molar concentration of A1M/hemopexin may be developed as a novel prognostic biomarker to assess AKI risk in SCD patients. We also provide proof of principle for hemopexin replacement therapy to prevent AKI in SCD during acute hemolytic events.
For original data, please e-mail the corresponding author.
Presented in part at the 59th and 61st annual meetings of the American Society of Hematology, Atlanta, GA, 9-12 December 2017, and Orlando, FL, 7-10 December 2019, respectively.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Soma Jobbagy for technical assistance with GFR measurements, and Akosua Osei-Tutu with TUNEL staining.
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute (1U54HL141011-01 and 1K01HL133331-01), and a 2019 Samuel and Emma Winters Foundation award.
Authorship
Contribution: S.F.O.-A. designed the study, provided clinical samples, and wrote the manuscript with S.G.; R.H. and O.O.O. performed biochemical experiments with human and mouse samples and analyzed data; D.C. performed BMT and GFR experiments; B.F. performed histological staining; D.L. performed biochemical experiments; E.B.A. collected and processed human samples; R.J.T. helped with histological scoring and reviewed the manuscript; D.A.V. performed GFR, analyzed data, and reviewed the manuscript; V.P. and E.O.-D. lead the ORDISS biorepository in Ghana; and S.G. conceived and designed the study, performed the experiments, analyzed data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the SickleGenAfrica Network appears in the supplemental data.
Correspondence: Samit Ghosh, University of Pittsburgh, BST-E1200-8B, 200 Lothrop St, Pittsburgh, PA 15261; e-mail: sag130@pitt.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal