

Abstract
Current objectives regarding treatment of acute myeloid leukemia (AML) include achieving complete remission (CR) by clinicopathological criteria followed by interrogation for the presence of minimal/measurable residual disease (MRD) by molecular genetic and/or flow cytometric techniques. Although advances in molecular genetic technologies have enabled highly sensitive detection of AML-associated mutations and translocations, determination of MRD is complicated by the fact that many treated patients have persistent clonal hematopoiesis (CH) that may not reflect residual AML. CH detected in AML patients in CR includes true residual or early recurrent AML, myelodysplastic syndrome or CH that is ancestral to the AML, and independent or newly emerging clones of uncertain leukemogenic potential. Although the presence of AML-related mutations has been shown to be a harbinger of relapse in multiple studies, the significance of other types of CH is less well understood. In patients who undergo allogeneic hematopoietic cell transplantation (HCT), post-HCT clones can be donor-derived and in some cases engender a new myeloid neoplasm that is clonally unrelated to the recipient’s original AML. In this article, we discuss the spectrum of CH that can be detected in treated AML patients, propose terminology to standardize nomenclature in this setting, and review clinical data and areas of uncertainty among the various types of posttreatment hematopoietic clones.
Introduction
Clonal hematopoiesis (CH), detectable populations of blood or marrow cells originating from somatically mutated hematopoietic stem or progenitor cell(s), frequently accompanies aging and is presumed to represent a precursor state for many hematologic neoplasms. The presence of CH with a mutant variant allele fraction (VAF) of at least 2% in individuals without cytopenias or a history of a myeloid neoplasm (ie, CH of indeterminate potential [CHIP]) is associated with an increased risk of developing a hematologic malignancy of 0.5% to 1% per year.1 Recent studies have shown that the presence of CHIP in cytopenic patients (ie, clonal cytopenia of undetermined significance) with high VAF (>10%) and specific mutation patterns is associated with a particularly high incidence of progression to a hematologic neoplasm.2,3 Additionally, individuals with CHIP are at higher risk for morbidity and mortality from nonneoplastic causes, including cardiovascular disease.4-6 The most common CHIP-associated mutations are also prevalent in myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), and other myeloid neoplasms. However, the significance of CH in patients treated for myeloid neoplasms is less well understood. In an AML patient in complete remission (CR), the persistence of mutations found at the time of AML diagnosis could reflect persistent leukemia with impending relapse, reversion to residual CHIP that preceded the AML diagnosis and that might pose only a small risk for leukemia recurrence, or even a separate clonal process unrelated to the AML.
In AML, recent studies have underscored the significance of minimal/measurable residual disease (MRD) detected by either flow cytometry immunophenotyping or sensitive molecular genetic techniques that interrogate for AML-associated mutations.7 MRD negativity is emerging as an alternative therapeutic end point that supplements and may eventually replace standard AML CR criteria, which rely on the morphologic detection of blasts and recovery of blood counts after therapy.7 However, although blast percentage has traditionally been considered as a proxy for AML disease burden, AML cases often have complex genetic hierarchies with variable association with blast count. In the posttreatment period, it may not be clear which mutations reflect the true AML cell burden and which may reflect a precursor CH. Additionally, mutations in several genes (such as DDX41, CEBPA, and RUNX1) can be either somatically acquired in the AML clone or occur as germline mutations that predispose to the development of myeloid neoplasia8 ; in the latter scenario, these mutations will persist at high VAF after therapy, even if the AML clone has been eliminated. Thus, the detection of mutations from the original AML in CR following therapy may have variable implications with respect to likelihood of disease recurrence. Moreover, there is a lack of consensus terminology for the varying patterns of detectable mutations in treated AML, which impedes study of their clinical implications.
Here, we review the types of CH that occur after treatment of AML, including molecular markers of true residual AML as well as states of CH that may not reflect persistent leukemia. We propose terminology to express these distinct types of CH to facilitate study of the management of AML patients posttherapy.
Current criteria for CR and relapsed disease in AML
Current International Working Group (IWG) and European LeukemiaNet (ELN) criteria for a CR following therapy for AML include achievement of a morphologic leukemia-free state (<5% blasts in the bone marrow [BM], no Auer rods, and no evidence of extramedullary disease) and peripheral blood count recovery (absolute neutrophil count >1 × 109/L and platelets >100 × 109/L, in the absence of growth factor treatment).9,10 Recurrent disease is defined as the reappearance of leukemic blasts in the blood or ≥5% leukemic blasts in the BM or the development of cytologically proven extramedullary disease; in IWG criteria, the appearance of “new dysplastic changes” is also considered to represent disease recurrence.9 These criteria continue to represent the backbone of treatment response determination in the routine care of AML patients as well as in clinical trials.
Despite broad use of these criteria, morphologic CR is an incomplete reflection of the disease state posttreatment. In some patients with ≥5% blasts after treatment, blasts may be shown by immunophenotype to be regenerative rather than neoplastic in nature, whereas conversely, many patients who attain CR harbor persistent low-level disease detectable by conventional karyotype, molecular genetics, or flow cytometry.11 Accumulating data have shown that the presence of such MRD in morphologic CR identifies patients at higher risk of relapse and who may benefit from additional therapy or more intensive therapy as opposed to those who have undetectable MRD.7
Molecular genetic interrogation of AML posttherapy
Techniques used to detect genetic aberrations in AML include conventional karyotype, fluorescent in situ hybridization (FISH), polymerase chain reaction (PCR)-based approaches (including real-time quantitative PCR [qPCR] and digital PCR), and next-generation sequencing (NGS)-based approaches. These methods vary widely in their applicability and sensitivities. Although cytogenetics and FISH can occasionally reveal evidence of MRD in morphologic CR,12 their sensitivities are generally similar to that of morphology (0.5 × 10−1), whereas a sensitivity of at least 1 × 10−3 is recommended for the detection of MRD.7 Chimerism studies can be used in the post–hematopoietic cell transplantation (HCT) setting to assess for the presence of recipient nonlymphoid hematopoietic cells (which are usually neoplastic and imply early recurrent AML), but are insensitive.13 Standard NGS, without the use of molecular barcoding, is limited by intrinsic sequencing error rates, with sensitivities of only 1 × 10−2 to 2 × 10−2.14 Nevertheless, the detection of AML-associated mutations even at the less-sensitive levels of 10−2 achievable in most clinical NGS platforms strongly predicts outcome in patients in morphologic CR.15,16 Newer technologies using molecular barcoding and error correction enable more sensitive detection of low-level mutation burden.
There is a wide spectrum of genetic abnormalities associated with AML and most cases bear several simultaneous abnormalities with distinct implications (Table 1). Genetic aberrations that occur late in AML ontogeny or characterize certain genetically defined AML subtypes are closely associated with the leukemic disease burden and have been shown to be markers of MRD. Conversely, mutations occurring early in AML ontogeny, in CHIP, or in preleukemic disease such as MDS, may not confer the same increased risk of relapse if found in the posttreatment setting. Some mutations can occur either early or late in AML ontogeny and have uncertain implications if found in the posttreatment setting.
The significance of posttherapy persistence of genetic abnormalities commonly seen in AML
| Genetic abnormality | Type | Techniques for detection | Usually cleared after successful therapy | Persistence after therapy associated with adverse outcome |
|---|---|---|---|---|
| RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA | AML-related | qPCR | Yes | Yes |
| NPM1 | AML-related | qPCR | Yes | Yes |
| KMT2A rearrangement, DEK-NUP214, BCR-ABL1 | AML-related | qPCR | Unknown | Unknown |
| NRAS/KRAS | AML-related | NGS | Yes | Yes |
| FLT3-ITD/FLT3-TKD | AML-related | NGS | Yes (but may be lost at relapse or acquired at relapse of previously FLT3 wild-type AML) | Unknown |
| PCR | ||||
| KIT | AML-related | NGS | Yes | Yes |
| PCR | ||||
| PTPN11 | AML-related | NGS | Yes | Yes |
| GATA2 | Likely AML-related | NGS | Yes | Unknown |
| CEBPA | Likely AML-related | NGS | Yes | Unknown |
| WT1 | Likely AML-related | NGS | Yes | Unknown |
| RUNX1 | CH (potentially AML-related) | NGS | Variable | Yes |
| IDH1/IDH2 | CH (potentially AML-related) | NGS | Variable | Yes |
| ddPCR | ||||
| DNMT3A | CH | NGS | Usually not | No |
| ASXL1 | CH | NGS | Variable | No |
| TET2 | CH | NGS | Usually not | No |
| SRSF2 | CH | NGS | Variable | No |
| BCOR | CH | NGS | Variable | No |
| TP53 | CH | NGS | Variable | Yes |
| Genetic abnormality | Type | Techniques for detection | Usually cleared after successful therapy | Persistence after therapy associated with adverse outcome |
|---|---|---|---|---|
| RUNX1-RUNX1T1, CBFB-MYH11, PML-RARA | AML-related | qPCR | Yes | Yes |
| NPM1 | AML-related | qPCR | Yes | Yes |
| KMT2A rearrangement, DEK-NUP214, BCR-ABL1 | AML-related | qPCR | Unknown | Unknown |
| NRAS/KRAS | AML-related | NGS | Yes | Yes |
| FLT3-ITD/FLT3-TKD | AML-related | NGS | Yes (but may be lost at relapse or acquired at relapse of previously FLT3 wild-type AML) | Unknown |
| PCR | ||||
| KIT | AML-related | NGS | Yes | Yes |
| PCR | ||||
| PTPN11 | AML-related | NGS | Yes | Yes |
| GATA2 | Likely AML-related | NGS | Yes | Unknown |
| CEBPA | Likely AML-related | NGS | Yes | Unknown |
| WT1 | Likely AML-related | NGS | Yes | Unknown |
| RUNX1 | CH (potentially AML-related) | NGS | Variable | Yes |
| IDH1/IDH2 | CH (potentially AML-related) | NGS | Variable | Yes |
| ddPCR | ||||
| DNMT3A | CH | NGS | Usually not | No |
| ASXL1 | CH | NGS | Variable | No |
| TET2 | CH | NGS | Usually not | No |
| SRSF2 | CH | NGS | Variable | No |
| BCOR | CH | NGS | Variable | No |
| TP53 | CH | NGS | Variable | Yes |
References: Schuurhuis et al,7 Morita et al,15 Jongen-Lavrencic et al,16 Wong et al,28 Ivey et al,29 Zhou et al,31 Venditti et al,32 Hourigan et al,33 Hirsch et al,38 Press et al,39 Höllein et al,41 Klco et al,42 Wong et al,47 Petrova et al,50 Debarri et al,51 Ok et al,53 Yilmaz et al,56 Gaidzik et al,72 Rothenberg-Thurley et al,73 and Thol et al.74
ddPCR, droplet digital PCR.
The AML-associated translocations RUNX1-RUNX1T1, CBFB-MYH11, and PML-RARA are highly specific for AML, and the presence of MRD for these markers in CR after therapy is predictive of relapse. Conversely, sensitive analysis that fails to detect MRD at the end of treatment is associated with excellent long-term relapse-free survival,17-19 although very low levels of CBFB-MYH11 and PML-RARA transcripts may persist in remission in a minority of cases.19-21 KMT2A and DEK-NUP214 translocations are also candidates for genetic aberrations closely associated with AML, although they are not currently considered to be AML defining.22-24 NPM1 mutation is strongly associated with AML, being rare in preleukemic myeloid neoplasms.25-27 NPM1-mutated cells decrease rapidly upon administration of induction chemotherapy,28 are usually undetectable in patients achieving durable CR,15 and reappear in almost all patients who relapse.29 Mutated NPM1 can be detected with high sensitivity by qPCR in both blood and BM, and MRD negativity for NPM1 after 2 cycles of consolidation predicts low risk of relapse,29,30 whereas the reappearance of NPM1 mutation in patients post-HCT is a strong predictor of subsequent morphologic relapse.31 In 1 study, preemptive treatment with azacitidine of patients in CR who developed detectable MRD for AML-associated translocations or mutated NPM1 was associated with relapse-free survival similar to patients lacking MRD.24 In another prospective study, patients with intermediate-risk AML who were selected to undergo HCT based on the MRD status of AML-associated genetic abnormalities had comparable overall survival to those without MRD.32 Furthermore, AML patients with detectable MRD appear to benefit from myeloablative rather than reduced-intensity conditioning when undergoing HCT.33 These results suggest that interrogation for the presence of MRD using AML-related genetic markers might not only be prognostic, but also help dictate specific therapeutic interventions.
The RAS signaling pathway genes FLT3, NRAS, and KRAS drive proliferation in myeloid neoplasia; activating mutations in these genes are associated with higher white blood counts and blast percentages.34-36 Most studies have shown that mutations in these genes (including both FLT3 internal tandem duplication [ITD] and tyrosine kinase domain [TKD] mutations) are relatively late events in AML evolution37 and do not persist in remission. Other mutations that are usually cleared following intensive AML therapy are KIT, PTPN11, WT1, as well as CEBPA and GATA2 (when the latter 2 mutations are somatic rather than germline).15,38-40 The detection of FLT3, NRAS, KRAS, PTPN11, or KIT mutations in CR is associated with a higher risk of relapse and shorter relapse-free survival.15,16,38,39,41,42
A central challenge in interpreting positive NGS results in the setting of AML CR is distinguishing true markers of residual AML from an AML precursor clone or an independent clone that emerges or expands after therapy.43 Unlike the AML-related genetic aberrations discussed in the preceding 2 paragraphs, DNMT3A, TET2, and ASXL1 (“DTA”) mutations occur early in AML evolution and are not alone sufficient to cause AML.44-46 They usually persist or even expand after intensive therapy that eliminates the AML-related mutations,28 but are eliminated following HCT with complete donor engraftment.29 This evidence suggests that DTA mutations should not be considered as markers of true residual AML and rather represent preleukemic CH, similar to CHIP mutations found in patients without a history of a hematologic neoplasm. Although there are less data than for DTA mutations, BCOR and SRSF2 mutations also appear to persist in most AML patients who achieve CR and are likely markers of CH.16,39 TP53 mutations can arise early or late in AML development, but are usually not cleared or even reduced following induction chemotherapy28 and may also emerge after therapy in a clone unrelated to the initial AML.47 The distinguishing features of AML-related and CH-type mutations are shown in Table 2.
AML-related vs CH-type genetic abnormalities
| AML-related genetic abnormalities | CH-type genetic abnormalities |
|---|---|
| • Often occur later in the mutation hierarchy; may be the sole detected genetic event | • Occur earlier in the mutation hierarchy, often at higher VAF in comparison to AML-related genetic abnormalities |
| • Reduction in VAF or clearance associated with a reduction in the blast percentage after therapy | • Often persist in CR, usually at similar VAF to the pretherapy disease |
| • Reappearance of genetic abnormality in relapsed disease | • Persistence in relapsed disease |
| • Presence in CR associated with increased risk of relapse | • Presence in CR may not be associated with increased risk of relapse |
| • Eliminated following successful HCT | • Eliminated following successful HCT |
| AML-related genetic abnormalities | CH-type genetic abnormalities |
|---|---|
| • Often occur later in the mutation hierarchy; may be the sole detected genetic event | • Occur earlier in the mutation hierarchy, often at higher VAF in comparison to AML-related genetic abnormalities |
| • Reduction in VAF or clearance associated with a reduction in the blast percentage after therapy | • Often persist in CR, usually at similar VAF to the pretherapy disease |
| • Reappearance of genetic abnormality in relapsed disease | • Persistence in relapsed disease |
| • Presence in CR associated with increased risk of relapse | • Presence in CR may not be associated with increased risk of relapse |
| • Eliminated following successful HCT | • Eliminated following successful HCT |
Aside from the AML-related and CH-type mutations discussed in the preceding 3 paragraphs, the implications of other mutations with respect to MRD are less clear. Their hierarchy is variable, and in different cases can reflect either pre-AML CH-type mutations or AML-related mutations.48,49 Unlike RAS pathway mutations, IDH1/IDH2 mutations frequently persist in CR,15,42 although usually at low levels (<2.5% VAF) that increase upon relapse.16,29,50 Detectable IDH1/IDH2 mutations in CR are associated with AML relapse, including in NPM1-comutated patients who clear the NPM1 mutation.51-53 Although RUNX1 mutation persistence in CR has been associated with shortened survival,54 it occurs frequently in pre-AML myeloid neoplasms such as MDS and often persists in CR after chemotherapy, features of a CH-type mutation.15,39 Thus, mutations other than the AML-related mutations and CH-type mutations can be considered as potentially, but not definitively, AML-related.
Proposed terminology to express post-AML CH states
The interpretation of genetic abnormalities in the posttherapy AML setting rests on the following information: (1) Was the posttreatment genetic abnormality present in the original AML (at a detectable level)? (2) Is the abnormality one of the AML-related mutations or gene rearrangements (Table 1)? (3) Is there morphologic evidence of a background MDS, myeloproliferative neoplasm (MPN), or MDS/MPN? An additional factor that may also impact the interpretation of mutational data is the VAF of the mutation(s) posttreatment in relation to their VAF at the time of diagnosis. The specific terms we propose are summarized and defined in Table 3.
Proposed terminology for post-AML CH states
| Term | Definition and significance |
|---|---|
| gMRD | AML-related genetic abnormality detectable after treatment (Figure 1A) |
| CH | Non-AML–related somatic genetic abnormality detectable after treatment, which may or may not have been detectable in the original diagnostic AML sample (Figure 1B-C) |
| Donor-derived CHIP | CH shown to be of donor hematopoietic cell origin, detected after HCT for AML (Figure 1I) |
| RMN | Morphologic and clinical evidence of MDS, MPN, or MDS/MPN in CR, supported by genetic evidence of clonality (CH) sharing any somatic genetic abnormalities with the antecedent AML (Figure 1D) |
| New myeloid neoplasm clonally unrelated to AML | MDS, MPN, or MDS/MPN with genetic features that are entirely different from the antecedent AML; if MDS or MDS/MPN, considered to be a therapy-related myeloid neoplasm |
| Donor-derived myeloid neoplasm | MDS, MPN, or MDS/MPN shown to be of donor hematopoietic cell origin, developing after HCT for AML |
| Germline mutation | Germline (nonsomatic) mutation, present in both pre- and posttherapy time points; should specify if donor-derived in the post-HCT setting (Figure 1E) |
| Recurrent/relapsed AML | ≥5% blasts in BM after CR with at least 1 AML-related and/or CH somatic genetic abnormality shared with the original AML (Figure 1F-G) |
| New clonally unrelated AML | ≥20% myeloid blasts* in blood or BM after CR lacking any shared somatic genetic aberrations with the original AML; considered to represent a therapy-related AML (Figure 1H) |
| Donor-derived AML | ≥20% myeloid blasts* in blood or BM shown to be of donor hematopoietic cell origin, developing after HCT for AML |
| Term | Definition and significance |
|---|---|
| gMRD | AML-related genetic abnormality detectable after treatment (Figure 1A) |
| CH | Non-AML–related somatic genetic abnormality detectable after treatment, which may or may not have been detectable in the original diagnostic AML sample (Figure 1B-C) |
| Donor-derived CHIP | CH shown to be of donor hematopoietic cell origin, detected after HCT for AML (Figure 1I) |
| RMN | Morphologic and clinical evidence of MDS, MPN, or MDS/MPN in CR, supported by genetic evidence of clonality (CH) sharing any somatic genetic abnormalities with the antecedent AML (Figure 1D) |
| New myeloid neoplasm clonally unrelated to AML | MDS, MPN, or MDS/MPN with genetic features that are entirely different from the antecedent AML; if MDS or MDS/MPN, considered to be a therapy-related myeloid neoplasm |
| Donor-derived myeloid neoplasm | MDS, MPN, or MDS/MPN shown to be of donor hematopoietic cell origin, developing after HCT for AML |
| Germline mutation | Germline (nonsomatic) mutation, present in both pre- and posttherapy time points; should specify if donor-derived in the post-HCT setting (Figure 1E) |
| Recurrent/relapsed AML | ≥5% blasts in BM after CR with at least 1 AML-related and/or CH somatic genetic abnormality shared with the original AML (Figure 1F-G) |
| New clonally unrelated AML | ≥20% myeloid blasts* in blood or BM after CR lacking any shared somatic genetic aberrations with the original AML; considered to represent a therapy-related AML (Figure 1H) |
| Donor-derived AML | ≥20% myeloid blasts* in blood or BM shown to be of donor hematopoietic cell origin, developing after HCT for AML |
The 20% blast threshold is applied for a genetically unrelated AML because this is a new disease and thus should fulfill criteria to establish a primary diagnosis of AML.
Genetic measurable residual disease
We propose the term genetic measurable residual disease (gMRD) to denote the presence of genetic abnormalities persisting or recurring after treatment that were present in the original AML and are AML related (Figure 1A). These include mutations or translocations that are much more frequent in AML than in non-AML myeloid neoplasms or CHIP, that generally disappear after successful intensive therapy, and the continued presence of which in CR has been shown to associate strongly with relapse. gMRD includes persistence of the AML-related genetic aberrations recommended to be monitored by the ELN due to the known clinical utility of MRD assessment in the posttreatment setting: mutations in NPM1 and RUNX1-RUNX1T1, CBFB-MYH11, and PML-RARA rearrangements. In addition, we propose that somatic KRAS, NRAS, FLT3 (both ITD and TKD), KIT (when not associated with underlying systemic mastocytosis), and PTPN11 mutations also be considered to be AML-related because there is evidence that those mutations are eliminated by successful intensive therapy and are associated with AML relapse when they persist. Although MRD-assessment data on DEK-NUP214 and KMT2A rearrangements are lacking, they are strongly associated with AML and we propose that they should be considered within the group of AML-related genetic aberrations. Somatic mutations in CEBPA, GATA2, and WT1 are also likely AML-related mutations, but there are less supporting data. The detection of germline mutations (GATA2, CEBPA, DDX41, and others) is not equivalent to gMRD. Following ELN guidelines for the reporting of MRD in AML, the significance of absent gMRD posttherapy depends on the sensitivity of detection: a lack of detectable gMRD should not be called “MRD absent” unless a highly sensitive detection technique (at least 10−3) is used. Most current NGS methods do not achieve this level of sensitivity and the thresholds of detection by NGS vary across genes. Using a sensitive technique such as qPCR, a complete molecular remission is defined as 2 successive gMRD-absent samples obtained at least 4 weeks apart, in the setting of a morphologic CR.7 According to recent ELN criteria, molecular genetic relapse is defined as an increase of the MRD level of at least 10-fold (1 log) between 2 positive samples in a patient who previously had undetectable MRD,7 even if criteria for morphologic recurrence are not met.
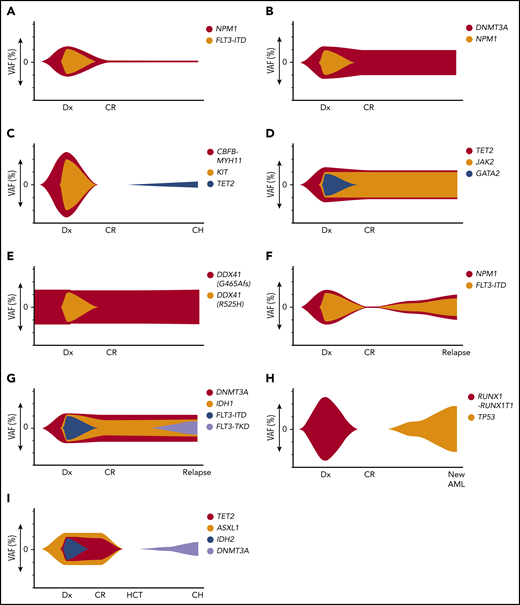
Illustrations of dynamic patterns of the mutational landscape after therapy for AML. (A) gMRD. At diagnosis (Dx), FLT3-ITD and NPM1 mutations were present. In CR, there is detectable NPM1 mutation, indicating the presence of gMRD despite morphologic CR. (B) CH that was detectable in the original AML. At diagnosis, DNMT3A and NPM1 mutations were present, both at high VAFs. In CR, there is no detectable NPM1 mutation, but a persistent DNMT3A mutation, indicating the presence of CH shared with the AML clone. If absence of NPM1 mutation is confirmed by a sensitive method, this would represent CH without gMRD. (C) CH that was not detected in the original AML. At diagnosis, CBFB-MYH11 rearrangement and KIT mutations were present, which disappeared in the initial CR time points. Subsequently, a TET2 mutation developed, indicating CH that was either present at a very low level prior to treatment (presumably not in the AML clone) or emerged after therapy. (D) RMN. At diagnosis, JAK2, TET2, and GATA2 mutations were present. The JAK2 and TET2 mutations persist in CR and BM shows morphologic features of primary myelofibrosis, indicating RMN despite morphologic CR and disappearance of the GATA2 mutation (which is likely AML-related in this case). (E) Persistent germline mutation. At diagnosis, 2 DDX41 mutations were present, 1 of which was shown to be of germline origin. After treatment in CR, the germline DDX41 mutation persists, with a similar VAF at all time points. (F) Recurrent AML. At diagnosis, NPM1 and FLT3-ITD mutations were present, which disappeared in CR. The same NPM1 and FLT3-ITD mutations are present at the time of relapse. (G) Recurrent AML with AML-related CH-type mutations, but new AML-related mutation. At diagnosis, DNMT3A, IDH1, and FLT3-ITD mutations were present, with persistence of the DNMT3A and IDH1 mutations during CR. At relapse, a new FLT3-TKD mutation has been acquired; nevertheless, the shared unique DNMT3A and IDH1 mutations indicate recurrence of the original clone rather than a new, unrelated clone. (H) New clonally unrelated AML. At diagnosis, RUNX1-RUNX1T1 rearrangement was present, which disappeared in CR. At relapse, there is a new TP53 mutation and a complex karyotype, but no RUNX1-RUNX1T1 rearrangement, indicating a new genetically distinct AML that is considered to be therapy-related. (I) Donor-derived CHIP. At diagnosis, TET2, ASXL1, and IDH2 mutation were present. Following HCT from an unrelated donor, the TET2, ASXL1, and IDH2 mutations disappear and the patient remains in CR, but a new DNMT3A mutation has appeared that was absent in the original leukemia. Chimerism studies show 100% donor chimerism, indicating DNMT3A-mutated CHIP that is donor derived (rather than originating from a low-level undetectable mutation that was present in the original recipient AML).
Illustrations of dynamic patterns of the mutational landscape after therapy for AML. (A) gMRD. At diagnosis (Dx), FLT3-ITD and NPM1 mutations were present. In CR, there is detectable NPM1 mutation, indicating the presence of gMRD despite morphologic CR. (B) CH that was detectable in the original AML. At diagnosis, DNMT3A and NPM1 mutations were present, both at high VAFs. In CR, there is no detectable NPM1 mutation, but a persistent DNMT3A mutation, indicating the presence of CH shared with the AML clone. If absence of NPM1 mutation is confirmed by a sensitive method, this would represent CH without gMRD. (C) CH that was not detected in the original AML. At diagnosis, CBFB-MYH11 rearrangement and KIT mutations were present, which disappeared in the initial CR time points. Subsequently, a TET2 mutation developed, indicating CH that was either present at a very low level prior to treatment (presumably not in the AML clone) or emerged after therapy. (D) RMN. At diagnosis, JAK2, TET2, and GATA2 mutations were present. The JAK2 and TET2 mutations persist in CR and BM shows morphologic features of primary myelofibrosis, indicating RMN despite morphologic CR and disappearance of the GATA2 mutation (which is likely AML-related in this case). (E) Persistent germline mutation. At diagnosis, 2 DDX41 mutations were present, 1 of which was shown to be of germline origin. After treatment in CR, the germline DDX41 mutation persists, with a similar VAF at all time points. (F) Recurrent AML. At diagnosis, NPM1 and FLT3-ITD mutations were present, which disappeared in CR. The same NPM1 and FLT3-ITD mutations are present at the time of relapse. (G) Recurrent AML with AML-related CH-type mutations, but new AML-related mutation. At diagnosis, DNMT3A, IDH1, and FLT3-ITD mutations were present, with persistence of the DNMT3A and IDH1 mutations during CR. At relapse, a new FLT3-TKD mutation has been acquired; nevertheless, the shared unique DNMT3A and IDH1 mutations indicate recurrence of the original clone rather than a new, unrelated clone. (H) New clonally unrelated AML. At diagnosis, RUNX1-RUNX1T1 rearrangement was present, which disappeared in CR. At relapse, there is a new TP53 mutation and a complex karyotype, but no RUNX1-RUNX1T1 rearrangement, indicating a new genetically distinct AML that is considered to be therapy-related. (I) Donor-derived CHIP. At diagnosis, TET2, ASXL1, and IDH2 mutation were present. Following HCT from an unrelated donor, the TET2, ASXL1, and IDH2 mutations disappear and the patient remains in CR, but a new DNMT3A mutation has appeared that was absent in the original leukemia. Chimerism studies show 100% donor chimerism, indicating DNMT3A-mutated CHIP that is donor derived (rather than originating from a low-level undetectable mutation that was present in the original recipient AML).
Flow cytometry can detect abnormal blast phenotypes at high levels of sensitivity and thus is an alternative technology for detecting MRD. Flow cytometry–detected MRD correlates reasonably well with gMRD detected by sensitive molecular genetic techniques, but shows discordant results in a subset of cases. Risk stratification appears to be optimized by using both methods simultaneously, with the highest risk of relapse in patients with both gMRD and immunophenotypic evidence of MRD.16,31,32,55
CH
We propose the term CH to denote the presence of any non-AML–related mutation or cytogenetic aberration in a patient in morphologic CR after AML treatment, without diagnostic morphologic features of a myeloid neoplasm (Figure 1B). The implications of CH in the post-AML setting may be distinct from CHIP detected in patients without an AML history, and are still not fully understood. Although persistent DTA mutations as sole aberrancies have not been shown to be associated with increased risk of relapse in some studies (with median time of follow-up of a few years), other studies suggest a long-term increased risk of recurrent AML.42,56 The presence of 2 or more CH-type mutations appears to predict a higher risk of relapse than a single mutation.38 Moreover, persistent DNMT3A-mutated hematopoiesis in NPM1-mutated AML patients in CR can engender the development of either NPM1− or NPM1+ relapsed AML, indicating that CH can provide a background for the emergence of a clonally related but genetically distinct relapse.41 The implications of the VAF and dynamic changes in VAF over time of these CH mutations are unknown; until further data are accumulated, specifying the genes and their VAFs in relation to the original AML is recommended. Chemotherapy, including AML-type chemotherapy, selects for chemoresistant clones that progressively expand and can confer risk for development of a subsequent myeloid neoplasm.47 Alternatively, novel CHIP clones could emerge as a consequence of patient aging (Figure 1C). However, relative VAF levels may not clearly establish the mutation hierarchy using current NGS methods, due to potential skewed amplification affecting the VAF and the inability to assign mutations to specific cell subpopulations in bulk-sequenced marrow or blood. Single-cell sequencing methods are more informative in this regard, but are currently not available for widespread clinical application.57
Aside from potential risk of AML relapse, CH mutations such as TET2 may confer increased risk of cardiovascular and other non-AML mortality, as has been shown in patients without history of hematologic malignancy.5,58 Persistent DNMT3A, TET2, ASXL1, or SRSF2 mutations (at VAF >20%) have also been associated with delayed peripheral blood count recovery following induction chemotherapy for AML.59 Further study is needed to determine the optimal management of CH in the post-AML setting.
Residual myeloid neoplasm and new clonally unrelated myeloid neoplasm
AML patients who meet the <5% blast count criterion for CR and lack AML-related mutations may have morphologic evidence of a background myeloid neoplasm and peripheral blood count abnormalities that do not appear to be related to residual effects of therapy or other exogenous factors, suggesting the presence of a background residual myeloid neoplasm (RMN). Many such patients have an antecedent history of MDS, MDS/MPN, or an MPN, but background myeloid neoplasia may also be “unmasked” by induction chemotherapy in a patient without a prior hematologic history (Figure 1D).60 These cases will usually have genetic aberrations typical of the myeloid neoplasm, such as JAK2, CALR, or MPL mutation in an underlying MPN; it is the morphologic and hematologic abnormalities that distinguish RMN from CH after AML. Of note, some cytogenetic aberrations, such as del(20q) or loss of the Y chromosome, are not MDS-defining and should be designated as CH unless there is morphologic evidence of a myeloid neoplasm.61 Conversely, morphologic abnormalities such as significant dysplasia and cytopenias should be interpreted with caution in the absence of supportive genetic evidence of a myeloid neoplasm. We propose that RMN should be diagnosed in patients treated for AML who are in remission from AML, but have compelling morphologic and clinical features of a myeloid neoplasm and supporting genetic findings. It is uncertain whether RMN has higher likelihood of AML relapse compared with CH without morphologic evidence of an underlying myeloid neoplasm, but it is recommended that these findings be reported to allow further study of this distinction. When reporting RMN, care must be taken to exclude residual effects of therapy or a transient exogenous factor such as a drug or infection, which may cause cytopenias and morphologic dysplasia. Splicing gene mutations and STAG2 mutations are uncommon in CHIP, thus their presence combined with morphologic dysplasia and cytopenias would tend to support RMN (MDS or MDS/MPN). Similarly, the presence of persistent JAK2, CALR, or MPL mutations would support a diagnosis of background MPN. As with CH, the specific genes and their VAFs in comparison with the original AML should be reported in the setting of RMN.
If a myeloid neoplasm emerges with a genetic profile that is entirely different from the original AML (with no shared mutations or cytogenetic aberrations), we propose that it be reported as a new, clonally unrelated myeloid neoplasm. According to the WHO classification, such cases displaying features of MDS or MDS/MPN would be considered therapy-related disease.62
Germline mutations
Certain germline mutations confer increased risk of developing AML and other myeloid neoplasms8 ; these mutations may be known prior to the patient’s AML or may be revealed by NGS at the time of AML diagnosis. Germline mutations will be detectable in all posttherapy time points in affected patients, with the exception of post-HCT using a donor lacking the germline mutation (Figure 1E). These mutations should be reported in posttherapy samples and designated as germline if known. Optimally, their nonsomatic origin should be proven by testing a nonhematopoietic tissue sample from the patient. In the post-HCT state, germline mutations originating from the donor marrow that were not present in the recipient should be reported as donor-derived germline mutations.
Recurrent/relapsed AML and new therapy-related AML
Relapsed AML is defined as the identification at least 5% blasts in the BM (or unexplained circulating blasts) following CR.9 If possible, clonal relationship to the prior AML should be proven by showing at least 1 shared mutation and/or cytogenetic aberration between the original and relapsed disease (Figure 1F). Significant genetic alterations from the original AML can be seen in relapsed disease, including gains and/or losses of mutations and cytogenetic aberrations.63 Any such changes should be reported, as mutations amenable to a targeted therapy may emerge or disappear in the relapsed disease.64 In some cases, only underlying CH-type mutations are shared between the original and relapsed AML, with loss of the original AML-related mutations and development of mutations that were not previously detected (Figure 1G).41,56,65 Although in effect this represents a clonally distinct disease, due to the shared background, classifying these cases as relapsed disease rather than a new clone is recommended.
Some putative relapsed AML cases are shown by cytogenetics and/or NGS to have a genetic profile completely unrelated to the original AML (Figure 1H). We propose that such cases be diagnosed as a “new AML” (therapy-related AML according to World Health Organization [WHO] classification guidelines) in distinction from relapsed disease that shares genetic features with the original AML.66 This distinction could be relevant in inclusion criteria for clinical trials targeting patients with relapsed AML vs therapy-related disease. These genetically unrelated secondary AML cases more commonly occur late (>2-3 years from the original AML diagnosis) and may be misclassified as relapsed disease, underscoring the need for repeat genetic analysis of recurrent AML, especially after a long latency.67 We propose that, unlike relapsed disease (which requires a lower blast threshold),9 a new AML should fulfill the same criteria for a primary AML diagnosis: at least 20% blasts in the blood or BM, and/or an AML-defining cytogenetic abnormality.
Donor-derived CHIP and donor-derived myeloid neoplasms
In the post-HCT setting, CH of donor origin may be detected, which in most cases represents CHIP that was present in the donor prior to the HCT68 and may confer an increased risk of hematologic neoplasia in the post-HCT patient.5 Correlation with BM chimerism studies is required to distinguish donor-derived CHIP from a recipient-derived CH. Similarly, donor-derived myeloid neoplasms such as MDS or AML may arise, which in some cases originate from CHIP already present in the donor stem cells or subsequently developed in the donor stem cells after HCT.69,70 Donor origin can be proven by the presence of 100% (or near 100%) donor chimerism and a genetic profile unrelated to the recipient’s original AML (Figure 1I). Any new CH or emerging myeloid neoplasm in the post-HCT setting that has a mutation pattern different from the original AML should be investigated for recipient vs donor origin, and we propose that donor-derived clones be designated as such.
Limitations
There are a number of factors that limit interpretation of results of the post-AML genetic profile compared with the original AML. Although some genetic aberrations are AML-related and can be used to track the presence or absence of AML, the relationship of other mutations, such as IDH1/2 and RUNX1, to the leukemic cell burden is uncertain. Moreover, not all patients have the AML genetic aberrations listed in Table 1 and it is uncertain how to interpret the significance of other, less common genetic aberrations that persist after AML therapy. If a mutation is cleared after therapy and reappears at a later time point during CR, this likely represents an AML-related mutation that is a harbinger of relapse. However, at the current time, only a limited set of AML-related genes have sufficient data to permit evidence-based treatment decisions based on the presence or absence of gMRD. We propose the terms “AML-related” and “CH” to qualify mutations in the post-AML setting with the realization that the list of genes assigned to these categories may change and categories could be further refined as additional evidence accumulates. Distinction between CH and a residual myeloid neoplasm can be particularly difficult given that both dysplasia and cytopenias are common in the post-AML setting, and in many cases this distinction may not be possible.
Detection of some gMRD lesions marked by chromosomal rearrangements can be achieved by reverse transcription–PCR, conventional karyotype, or FISH, albeit with low sensitivity. However, sensitive gMRD detection technologies are currently not available in most centers or are only available for selected genes and selected hotspot mutations. Clinical NGS testing can successfully detect the presence of the common AML-related and CH-type mutations to the level of ∼10−2, which is more sensitive than both morphologic assessment for increased blasts and gMRD detected by conventional karyotype or FISH, but does still not reach the threshold of 10−3 recommended to define MRD absence. Also, NGS sensitivity can be variable depending on the specific genetic locus. Thus, although the presence of persistent AML-related mutations detected by NGS can be taken as evidence of gMRD, the failure to detect these mutations on most NGS-based panels is insufficiently sensitive to confirm an MRD-absent state. Entry of more sensitive NGS technologies into the clinical testing arena will foster more widespread applicability of NGS for detecting gMRD.
Finally, patients with AML are increasingly being treated with therapies other than intensive induction chemotherapy (eg, DNA-hypomethylating agents combined with venetoclax or other agents, or targeted therapies), and genetic responses to these nonintensive therapies could differ from responses seen after induction. Most studies have focused on the dynamics of mutation clearance and the significance of mutation persistence after intensive induction chemotherapy and HCT, but very few studies have examined changes in mutation profiles after hypomethylating agents or targeted therapies. Responses of both AML-related and CH-type mutations may vary according to the type of therapy: for example, 1 study found that AML-related mutations persisted at low VAFs for 1 year or longer in some patients in continuous CR following hypomethylating agent therapy.71 Thus, analysis of the significance of post-AML mutation patterns may need to be tailored to the specific type of therapy administered.
Conclusions
With the increased interrogation of the genetic landscape of AML following therapy and the advent of highly sensitive genetic interrogation methods comes increased complexity in the interpretation of results. Accumulating evidence suggests that, although some genetic aberrations truly reflect residual AML, mutations in many other genes reflect a state of CH. The former can serve as markers of AML MRD, whose presence informs prognosis and also could direct therapeutic intervention even in the setting of morphologic CR. The latter bear similarities to CHIP and are not considered equivalent to residual AML. The altered microenvironment of the post-AML therapy patient (including patients post-HCT) likely confers different implications of post-AML CH states compared with CHIP and will require further study to optimize management. In treated AML patients who undergo NGS and other genetic testing, we propose that uniform nomenclature be used in pathology reports and in the patients’ clinical records to reflect these different types of genetic disease states. Recognizing the different implications of mutation patterns post-AML therapy will facilitate further study of therapeutic intervention and optimal patient management according to each specific scenario. However, we acknowledge that this nomenclature may not be applicable to all cases due to current limitations in the sensitivity of genetic testing, the ability to construct a genetic hierarchy, and knowledge of the clinical implications of the genetic profile. Thus, these proposals should be considered as a starting point for discussion and are amenable to refinement as additional evidence accumulates.
Acknowledgments
The authors thank Vineet Sharma for assistance with figure preparation.
This work was supported by P50CA206963 from the National Institutes of Health, National Cancer Institute (D.P.S., T.A.G., B.L.E.). D.P.S. was supported by the Edward P. Evans Foundation and the James and Lois Champy Fund. B.L.E. is an investigator of the Howard Hughes Medical Institute.
Authorship
Contribution: R.P.H. designed research, analyzed data, and wrote the paper; and D.P.S., T.A.G., and B.L.E. designed research and analyzed data.
Conflict-of-interest disclosure: B.L.E. has received research funding from Celgene and Deerfield; has received consulting fees from GRAIL; and serves on the scientific advisory boards for, and holds equity in, Skyhawk Therapeutics and Exo Therapeutics. T.G. has received research funding from Janssen and Calico. The remaining authors declare no competing financial interests.
Correspondence: Robert P. Hasserjian, Department of Pathology, Massachusetts General Hospital, WRN244, 55 Fruit St, Boston, MA 02114; e-mail: rhasserjian@partners.org.
