

Key Points
IPS-E is a simple and robust prognostic model for early-stage CLL.
IPS-E can be helpful in patients’ counseling and design of clinical trials.

Abstract
Most patients with chronic lymphocytic leukemia (CLL) are diagnosed with early-stage disease and managed with active surveillance. The individual course of patients with early-stage CLL is heterogeneous, and their probability of needing treatment is hardly anticipated at diagnosis. We aimed at developing an international prognostic score to predict time to first treatment (TTFT) in patients with CLL with early, asymptomatic disease (International Prognostic Score for Early-stage CLL [IPS-E]). Individual patient data from 11 international cohorts of patients with early-stage CLL (n = 4933) were analyzed to build and validate the prognostic score. Three covariates were consistently and independently correlated with TTFT: unmutated immunoglobulin heavy variable gene (IGHV), absolute lymphocyte count higher than 15 × 109/L, and presence of palpable lymph nodes. The IPS-E was the sum of the covariates (1 point each), and separated low-risk (score 0), intermediate-risk (score 1), and high-risk (score 2-3) patients showing a distinct TTFT. The score accuracy was validated in 9 cohorts staged by the Binet system and 1 cohort staged by the Rai system. The C-index was 0.74 in the training series and 0.70 in the aggregate of validation series. By meta-analysis of the training and validation cohorts, the 5-year cumulative risk for treatment start was 8.4%, 28.4%, and 61.2% among low-risk, intermediate-risk, and high-risk patients, respectively. The IPS-E is a simple and robust prognostic model that predicts the likelihood of treatment requirement in patients with early-stage CLL. The IPS-E can be useful in clinical management and in the design of early intervention clinical trials.
Introduction
Chronic lymphocytic leukemia (CLL) is characterized by the relentless accumulation of monoclonal B lymphocytes with a distinct immunophenotype (ie, CD5, CD19, CD20, CD23) in peripheral blood, bone marrow, and lymphoid organs. CLL is the most frequent form of leukemia in Western countries, where 0.6% of the population is diagnosed with this B-cell tumor during their lifetime.1 In most instances, CLL is diagnosed in general practice, and approximately 70% of patients present in an early phase of the disease, with no anemia, no thrombocytopenia, and no significantly enlarged lymph nodes or spleen.2
According to international guidelines, patients with asymptomatic early-stage CLL should not be treated until disease progression occurs, and active surveillance remains the standard for management.3-5 Indeed, different clinical trials enrolling patients with early-stage CLL failed to show a survival benefit of early intervention strategies based on chemo-chemoimmunotherapy.6-10 However, patients with early-stage CLL have a variable clinical course. Some require treatment soon after diagnosis because of development of cytopenia or bulky lymphadenopathy, whereas others show a stable or a slowly progressive disease not requiring treatment of decades.11 Because of this, the management of patients with early-stage CLL is challenging and shaped by uncertainty. Of note, it is estimated that more than 400 000 individuals in Europe and the United States diagnosed with CLL are followed by active surveillance.1,12 Unfortunately, there is no a specific, simple, accurate, and widely accepted prognostic model to predict the likelihood of disease progression, and hence need for therapy, in patients with asymptomatic early-stage CLL.
The management of early, asymptomatic CLL may change in the future if a survival benefit is proven by early intervention with novel agents in patients who are at risk for impending progression to symptomatic disease requiring treatment.13 In this new scenario, upfront identification of high-risk patients is warranted. The objective of this study was to construct a simple, robust, and validated predictor of disease progression and the need for intervention in patients with asymptomatic early-stage CLL.
Methods
Study design
This is a multicenter, international, retrospective, observational study in which already-existing and coded health-related data were further used (ClinicalTrials.gov identifier: NCT03436524). We obtained the individual patient data sets from 11 cohorts, including 8 series that were previously used to generate or validate scoring systems in CLL (Tables 1 and 2).10,14-19 Inclusion criteria were: presentation after 1996 confirmed by flow cytometry3,4 ; Binet stage A at diagnosis, as defined by blood cell count and physical examination3,4 ; and active surveillance as initial management, defined by no treatment requirement within the first 3 months after diagnosis. The Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK) criteria were followed throughout the study.20 Patients provided informed consent in accordance with local institutional review board requirements and the Declaration of Helsinki. The ethics committee approved the study (BASEC 2018-00341). Early-stage CLL was defined according to the Binet system in 10 of 11 cohorts. Data of individual patients with early (Binet stage A) disease managed with active surveillance policy were collected from the CLL1 (ClinicalTrials.gov identifier: NCT00262782; N = 547)9,18 and CLL7 (ClinicalTrials.gov identifier: NCT00275054; N = 339) trials of the German CLL Study Group, and the O-CLL1 GISL trial (ClinicalTrials.gov identifier: NCT00917540; N = 312).15,19 The M.D. Anderson Cancer Center (N = 1225),14 the University of Eastern Piedmont (UEP; N = 333),17 the Barcelona (N = 355),19 the Brno University Hospital (Brno; N = 269),19 the Southampton (N = 226), and the Sapienza University (N = 223) cohorts are institutional consecutive series of newly presented and prospectively observed patients with early (Binet stage A) disease. Binet stage A patients’ data of the SCAN cohort (N = 223) were collected from the SCALE Scandinavian population-based case-control study.16 The Mayo Clinic cohort (N = 881) is an institutional series of patients with early stage 0, I, and II disease defined according to the Rai system.
Characteristics of the training cohort
| Variable | UEP (N = 333) | ||
|---|---|---|---|
| N | % | Missing | |
| Age >65 years | 208 | 62.4 | — |
| Male sex | 178 | 53.4 | — |
| Palpable lymph nodes | 68 | 20.4 | — |
| Palpable spleen | 20 | 6.0 | — |
| Lymphocytes >15 × 109/L | 65 | 19.5 | — |
| Hb <13 g/dL (male)/<12 g/dL (female) | 39 | 11.7 | — |
| Platelets <150 × 109/L | 46 | 13.8 | — |
| B2M >3.5 mg/L | 32 | 9.6 | — |
| FISH normal | 112 | 33.6 | — |
| Del(13q) | 168 | 50.4 | — |
| Trisomy 12 | 59 | 17.7 | — |
| Del(11q) | 18 | 5.4 | — |
| Del(17p) | 19 | 5.7 | — |
| Unmutated IGHV | 92 | 27.6 | — |
| TP53 mutation | 24 | 7.2 | — |
| ATM mutation | 24 | 8.3 | 44 |
| MYD88 mutation | 18 | 5.4 | 4 |
| NOTCH1 mutation | 35 | 10.6 | 4 |
| SF3B1 mutation | 17 | 5.1 | 4 |
| CLL-IPI | |||
| Low risk | 182 | 54.7 | |
| Intermediate risk | 94 | 28.2 | |
| High risk | 45 | 13.5 | |
| Very high risk | 12 | 3.6 | |
| Variable | UEP (N = 333) | ||
|---|---|---|---|
| N | % | Missing | |
| Age >65 years | 208 | 62.4 | — |
| Male sex | 178 | 53.4 | — |
| Palpable lymph nodes | 68 | 20.4 | — |
| Palpable spleen | 20 | 6.0 | — |
| Lymphocytes >15 × 109/L | 65 | 19.5 | — |
| Hb <13 g/dL (male)/<12 g/dL (female) | 39 | 11.7 | — |
| Platelets <150 × 109/L | 46 | 13.8 | — |
| B2M >3.5 mg/L | 32 | 9.6 | — |
| FISH normal | 112 | 33.6 | — |
| Del(13q) | 168 | 50.4 | — |
| Trisomy 12 | 59 | 17.7 | — |
| Del(11q) | 18 | 5.4 | — |
| Del(17p) | 19 | 5.7 | — |
| Unmutated IGHV | 92 | 27.6 | — |
| TP53 mutation | 24 | 7.2 | — |
| ATM mutation | 24 | 8.3 | 44 |
| MYD88 mutation | 18 | 5.4 | 4 |
| NOTCH1 mutation | 35 | 10.6 | 4 |
| SF3B1 mutation | 17 | 5.1 | 4 |
| CLL-IPI | |||
| Low risk | 182 | 54.7 | |
| Intermediate risk | 94 | 28.2 | |
| High risk | 45 | 13.5 | |
| Very high risk | 12 | 3.6 | |
B2M, β-2-microglobulin; Del, deletion; Hb, hemoglobin.
Characteristics of the validation cohorts
| Variable | MDACC (N = 1225) | CLL1 (N = 547) | Barcelona (N = 355) | CLL7 (N = 339) | O-CLL1 (N = 312) | Brno (N = 269) | Southampton (N = 226) | SCAN (N = 223) | SU (N = 223) | Mayo Clinic (N = 881) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | |
| Age >65 years | 374 | 30.5 | — | 154 | 28.1 | — | 166 | 46.7 | — | 98 | 28.9 | — | 80 | 25.6 | — | 113 | 42.0 | — | 120 | 53.0 | — | 95 | 42.6 | — | 28 | 12.7 | 3 | 380 | 43.1 | — |
| Male sex | 727 | 59.3 | — | 335 | 61.2 | — | 202 | 56.9 | — | 214 | 63.1 | — | 190 | 60.8 | — | 167 | 62.0 | — | 134 | 59.2 | — | 131 | 58.7 | — | 124 | 55.6 | — | 591 | 67.1 | — |
| Palpable lymph nodes | 536 | 43.7 | — | 122 | 22.3 | — | 81 | 22.8 | — | 115 | 33.9 | — | 62 | 19.8 | — | 119 | 44.2 | — | 93 | 41.1 | — | 94 | 42.1 | — | 114 | 51.1 | — | 424 | 48.1 | — |
| Palpable spleen | 68 | 5.5 | — | 29 | 5.5 | 24 | 6 | 1.6 | — | 16 | 5.1 | 28 | 21 | 6.7 | — | 2 | 0.7 | — | 4 | 1.7 | — | 7 | 3.2 | 5 | 29 | 13.0 | — | 62 | 7.0 | — |
| Lymphocytes >15 × 109/L | 609 | 49.7 | — | 280 | 51.1 | — | 91 | 25.6 | — | 141 | 41.6 | 139 | 44.5 | — | 199 | 73.9 | — | 78 | 34.5 | — | 127 | 56.9 | — | 75 | 33.6 | — | 353 | 40.1 | 2 | |
| B2M >3.5 mg/L | 116 | 9.7 | 41 | 44 | 8.0 | — | 39 | 11.3 | 11 | 7 | 2.1 | 16 | 3 | 1.3 | 82 | 31 | 11.9 | 10 | 31 | 20.6 | 76 | 16 | 7.3 | 5 | 8 | 3.6 | 1 | 80 | 9.7 | 53 |
| Trisomy 12 | 196 | 16.0 | — | 49 | 9.1 | 12 | 55 | 16.5 | 23 | 28 | 8.3 | 3 | 26 | 9.2 | 30 | 31 | 11.5 | 1 | 20 | 10.9 | 44 | 12 | 6.9 | 51 | 20 | 10.4 | 31 | 151 | 17.8 | 33 |
| Del(17p) | 69 | 5.6 | — | 15 | 2.8 | 12 | 14 | 4.1 | 15 | 7 | 2.0 | 3 | 6 | 2.1 | 30 | 18 | 6.6 | — | 7 | 3.7 | 38 | 3 | 1.7 | 51 | 8 | 4.1 | 31 | 40 | 4.8 | 33 |
| Unmutated IGHV | 480 | 39.1 | — | 157 | 28.7 | — | 138 | 38.8 | — | 74 | 21.8 | — | 106 | 33.9 | — | 136 | 50.5 | — | 70 | 30.9 | — | 56 | 25.1 | — | 70 | 31.3 | — | 387 | 44.7 | 15 |
| CLL-IPI | 41 | 14 | 26 | 43 | 93 | 10 | 99 | 10 | 19 | 76 | ||||||||||||||||||||
| Low risk | 566 | 47.8 | 330 | 61.9 | 174 | 52.9 | 201 | 67.9 | 136 | 62.1 | 98 | 37.8 | 57 | 44.9 | 134 | 62.9 | 129 | 63.2 | 349 | 43.4 | ||||||||||
| Intermediate risk | 422 | 35.7 | 138 | 25.9 | 93 | 28.3 | 76 | 25.7 | 72 | 32.9 | 91 | 35.1 | 41 | 32.3 | 54 | 25.4 | 49 | 24.1 | 290 | 36.0 | ||||||||||
| High risk | 160 | 13.5 | 58 | 10.9 | 46 | 14.0 | 18 | 6.1 | 10 | 4.6 | 45 | 17.4 | 27 | 21.2 | 22 | 10.3 | 19 | 9.3 | 142 | 17.6 | ||||||||||
| Very high risk | 36 | 3.0 | 7 | 1.3 | 16 | 4.8 | 1 | 0.3 | 1 | 0.4 | 25 | 9.7 | 2 | 1.6 | 3 | 1.4 | 7 | 3.4 | 24 | 3.0 | ||||||||||
| Variable | MDACC (N = 1225) | CLL1 (N = 547) | Barcelona (N = 355) | CLL7 (N = 339) | O-CLL1 (N = 312) | Brno (N = 269) | Southampton (N = 226) | SCAN (N = 223) | SU (N = 223) | Mayo Clinic (N = 881) | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | N | % | Miss. | |
| Age >65 years | 374 | 30.5 | — | 154 | 28.1 | — | 166 | 46.7 | — | 98 | 28.9 | — | 80 | 25.6 | — | 113 | 42.0 | — | 120 | 53.0 | — | 95 | 42.6 | — | 28 | 12.7 | 3 | 380 | 43.1 | — |
| Male sex | 727 | 59.3 | — | 335 | 61.2 | — | 202 | 56.9 | — | 214 | 63.1 | — | 190 | 60.8 | — | 167 | 62.0 | — | 134 | 59.2 | — | 131 | 58.7 | — | 124 | 55.6 | — | 591 | 67.1 | — |
| Palpable lymph nodes | 536 | 43.7 | — | 122 | 22.3 | — | 81 | 22.8 | — | 115 | 33.9 | — | 62 | 19.8 | — | 119 | 44.2 | — | 93 | 41.1 | — | 94 | 42.1 | — | 114 | 51.1 | — | 424 | 48.1 | — |
| Palpable spleen | 68 | 5.5 | — | 29 | 5.5 | 24 | 6 | 1.6 | — | 16 | 5.1 | 28 | 21 | 6.7 | — | 2 | 0.7 | — | 4 | 1.7 | — | 7 | 3.2 | 5 | 29 | 13.0 | — | 62 | 7.0 | — |
| Lymphocytes >15 × 109/L | 609 | 49.7 | — | 280 | 51.1 | — | 91 | 25.6 | — | 141 | 41.6 | 139 | 44.5 | — | 199 | 73.9 | — | 78 | 34.5 | — | 127 | 56.9 | — | 75 | 33.6 | — | 353 | 40.1 | 2 | |
| B2M >3.5 mg/L | 116 | 9.7 | 41 | 44 | 8.0 | — | 39 | 11.3 | 11 | 7 | 2.1 | 16 | 3 | 1.3 | 82 | 31 | 11.9 | 10 | 31 | 20.6 | 76 | 16 | 7.3 | 5 | 8 | 3.6 | 1 | 80 | 9.7 | 53 |
| Trisomy 12 | 196 | 16.0 | — | 49 | 9.1 | 12 | 55 | 16.5 | 23 | 28 | 8.3 | 3 | 26 | 9.2 | 30 | 31 | 11.5 | 1 | 20 | 10.9 | 44 | 12 | 6.9 | 51 | 20 | 10.4 | 31 | 151 | 17.8 | 33 |
| Del(17p) | 69 | 5.6 | — | 15 | 2.8 | 12 | 14 | 4.1 | 15 | 7 | 2.0 | 3 | 6 | 2.1 | 30 | 18 | 6.6 | — | 7 | 3.7 | 38 | 3 | 1.7 | 51 | 8 | 4.1 | 31 | 40 | 4.8 | 33 |
| Unmutated IGHV | 480 | 39.1 | — | 157 | 28.7 | — | 138 | 38.8 | — | 74 | 21.8 | — | 106 | 33.9 | — | 136 | 50.5 | — | 70 | 30.9 | — | 56 | 25.1 | — | 70 | 31.3 | — | 387 | 44.7 | 15 |
| CLL-IPI | 41 | 14 | 26 | 43 | 93 | 10 | 99 | 10 | 19 | 76 | ||||||||||||||||||||
| Low risk | 566 | 47.8 | 330 | 61.9 | 174 | 52.9 | 201 | 67.9 | 136 | 62.1 | 98 | 37.8 | 57 | 44.9 | 134 | 62.9 | 129 | 63.2 | 349 | 43.4 | ||||||||||
| Intermediate risk | 422 | 35.7 | 138 | 25.9 | 93 | 28.3 | 76 | 25.7 | 72 | 32.9 | 91 | 35.1 | 41 | 32.3 | 54 | 25.4 | 49 | 24.1 | 290 | 36.0 | ||||||||||
| High risk | 160 | 13.5 | 58 | 10.9 | 46 | 14.0 | 18 | 6.1 | 10 | 4.6 | 45 | 17.4 | 27 | 21.2 | 22 | 10.3 | 19 | 9.3 | 142 | 17.6 | ||||||||||
| Very high risk | 36 | 3.0 | 7 | 1.3 | 16 | 4.8 | 1 | 0.3 | 1 | 0.4 | 25 | 9.7 | 2 | 1.6 | 3 | 1.4 | 7 | 3.4 | 24 | 3.0 | ||||||||||
Miss., missing.
Data analysis
We consecutively performed both uni- and multivariable analyses using the full data set of the training cohort (UEP). Subsequently, we analyzed the external validation data sets to confirm the findings from the full-analysis data set. The study end point was time to first treatment (TTFT), defined as the time between presentation and start of first treatment of CLL because of progression to symptomatic disease according to the National Cancer Institute-Working Group/International Workshop on Chronic Lymphocytic Leukemia guidelines (patients without a documented event were censored at the date of last observation or death).3,4 Nineteen baseline biomarkers, assessed within 1 month from initial presentation, were initially considered as covariates for construction of the prognostic index. These covariates were clinical characteristics (age, sex, ≥1 palpable lymph node with a diameter ≥1 cm and palpable spleen by investigator’s physical examination),21 laboratory values (hemoglobin level, platelet count, absolute lymphocyte count, and β-2-microglobulin), cytogenetic abnormalities as assessed by fluorescence in situ hybridization [FISH; del(17p), del(11q), trisomy 12, del(13q)], and gene mutations (immunoglobulin heavy variable gene [IGHV], ATM, MYD88, NOTCH1, SF3B1, and TP53). FISH was performed for assessing the t(11;14) in those cases in which a diagnosis of mantle cell lymphoma could be considered; patients harboring such translocations were excluded from the study. Continuous variables were categorized by published thresholds (age, hemoglobin, platelet count, β-2-microglobulin, IGHV identity)18,22-25 or by identifying the best cutoff through recursive partitioning (absolute lymphocyte count; supplemental Figure 1, available on the Blood Web site). Blood cell count was assessed by local laboratories. β-2-microglobulin was assessed by local laboratories with the exception of the CLL1, CLL7, and SCAN cohorts for which the information was provided centrally. FISH cytogenetics were assessed locally, with the exception of the CLL1, CLL7, and O-CLL1 cohorts, for which the information was provided centrally and scored according to the laboratory cutoff. IGHV and TP53 mutations were assessed by Sanger sequencing locally, with the exception of CLL1, CLL7, and O-CLL1 cohorts, for which the information was provided centrally. Mutations of ATM, MYD88, NOTCH1, and SF3B1 were assessed in the training cohort by targeted deep next-generation sequencing, using an allele frequency of at least 10% for defining a positive result.26 For the validation data sets, only variables composing the final model were included in the analyses. In the training and validation cohorts, minimal missing data were documented among covariates used for development and validation of the prognostic score. Therefore, no missing data imputation procedures were used. All statistical tests were 2-sided. Statistical significance was defined as P < .05. The analysis was performed with the Statistical Package for the Social Sciences software v.22.0 (Chicago, IL) and with R statistical package v3.4.1 (www.r-project.org).
Development and validation of a score for TTFT prognostication
Survival analysis was performed by the Kaplan-Meier method and comparison between strata, using the log-rank test.27 Sensitivity analyses of TTFT were performed by considering death without treatment as a competing risk and by using Gray’s test for comparisons between strata.28,29 The adjusted association between exposure variables and TTFT was estimated by Cox regression.30 In the training cohort, Cox regression included exposure variables showing an univariable association with TTFT with a Bonferroni corrected P < .05 to account for multiple testing. Backward elimination using likelihood ratio statistics with selection criterion P < .05 was used to derive the final Cox model to be validated. An elastic net penalized regression model was fit to the training set to exclude potential variable loss in the backward selection procedure.31 The mixing parameter was selected using 10-fold cross-validation. The ideal model was selected as that corresponding to the largest value of the tuning parameter, such that the error was within 1 standard error of the minimum mean cross-validated error. The proportional hazard assumption was assessed by plotting the smoothed Schoenfeld residuals against time.32 The stability of the Cox model was externally validated across the 9 Binet A validation cohorts. Only variables independently and significantly associated with TTFT in more than 50% of the validation cohorts were used to construct the final score. We assigned a weighted-risk score to each factor based on the regression parameters from the Cox regression analysis. The score was defined as the sum of single-risk parameters.18 We stratified patients in different risk groups by recursive partitioning of the score.33 Two major steps were used to derive the best decision tree: growing an initial tree under the following constraints and stopping rules: i) split criteria of P < .01 according to the log-rank test, ii) more than 20 patients in a node to be considered for splitting, and iii) more than 10 patients in a terminal node; and applying a pruning algorithm based on the complexity parameter (cp > 0.01). Model discrimination was computed using the Harrel’s C-index.34 The 9 Binet A validation cohorts were meta-analyzed to interpolate the validation series in a single plot. TTFT and corresponding 95% confidence intervals (95% CIs) were estimated in each of the Binet A validation cohorts at 6-month intervals. At each point, a random-effect model was used to provide a meta-analytic estimate of TTFT and corresponding 95% CIs across the Binet A validation cohorts. Survival curves were plotted by fitting a cubic smoothing spline to the estimates of TTFT and the respective 95% CIs, using a smoothing parameter of 0.6. A fixed-effect model was used for the meta-analysis of the C-index across the Binet A cohorts, the 1- and 5-year cumulative risk for treatment need, and number of events per 100 person years, and bootstrapping was used to calculate 95% CI. Heterogeneity was assessed by using the I2 statistic and Cochran’s Q test.
Results
Individual patient data from 11 cohorts of patients with early-stage CLL initially managed with active surveillance were collected, accounting for a total of 4933 patients (Tables 1 and 2). At the time of treatment, Binet stage and/or indication for therapy were consistent across the study cohorts and were similar to those reported in first-line clinical trials (supplemental Table 1), thus mitigating the risk for biases resulting from heterogeneity in treatment initiation triggers.
By univariable analysis in the UEP training series, which reflects patients managed in daily practice (N = 333; median follow-up, 7.2 years; number of treatment events, 95), baseline factors associated with an increased risk for treatment need (ie, TTFT) with a Bonferroni corrected P value < .05 were palpable lymph nodes, absolute lymphocyte count (>15 × 109/L), mild anemia (hemoglobin between 10 and 13 g/dL in men and 10 and 12 g/dL in women), unmutated IGHV genes, and trisomy 12 (Figure 1A). Sensitivity analysis accounting for death without treatment as competing risk confirmed the univariable associations between baseline variables and TTFT (supplemental Table 2). By multivariable analysis in the training series, 4 variables independently associated with TTFT were identified, including unmutated IGHV genes, absolute lymphocyte count higher than 15 × 109/L, palpable lymph nodes, and trisomy 12 (Figure 1B). The elastic net approach confirmed the stability of the selected variables, as the coefficients for unmutated IGHV genes, absolute lymphocyte count higher than 15 × 109/L, palpable lymph nodes, and trisomy 12 were not penalized and therefore not shrunk to zero. Unmutated IGHV genes, absolute lymphocyte count higher than 15 × 109/L, and palpable lymph nodes, but not trisomy 12, were found to be the most stable biomarkers after iterating the multivariable analysis in each single-validation Binet A cohort (Figure 1C). Across the validation cohorts, unmutated IGHV genes, absolute lymphocyte count higher than 15 × 109/L, and palpable lymph nodes maintained the highest percentage of selection in the final model, as well as after including in the multivariable analyses del(17p) and elevated β-2-microglobulin, which are part of the CLL-IPI score (Figure 1D).18
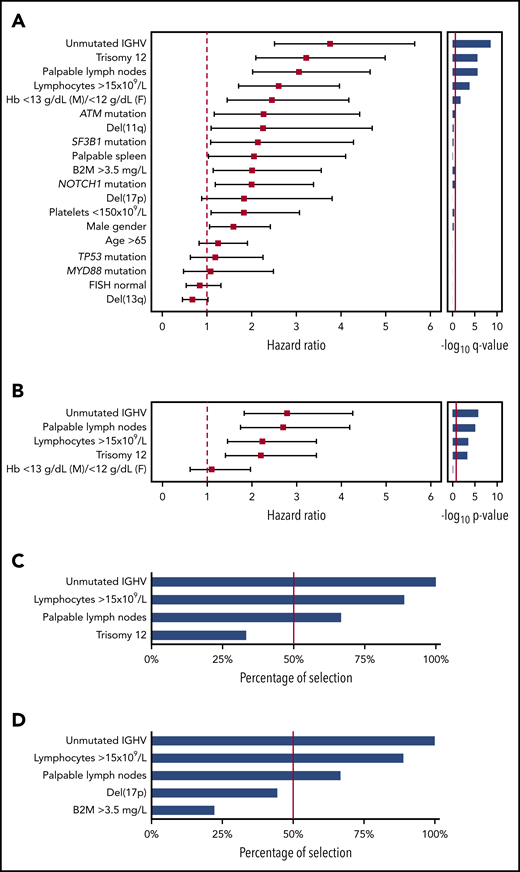
Univariable and multivariable analysis for time to first treatment. (A) Forest plot of the hazard ratio for the 19 covariates assessed for association with TTFT by univariable analysis. Solid boxes indicate the hazard ratio, horizontal lines indicate the 95% CIs. The red dot line marks hazard ratio of 1. The bar graph on the left shows the multiplicity adjusted -log10 P value by Bonferroni. The red solid line marks the .05 significance level. (B) Forest plot of the hazard ratio for the 5 covariates assessed for association with TTFT by multivariable analysis. Solid boxes indicate the hazard ratio, horizontal lines indicate the 95% CIs. The red dot line marks hazard ratio of 1. The bar graph on the left shows the -log10 P value. The red solid line marks the .05 significance level. (C-D) The bar plot shows the percentage of covariate selection in the final multivariable model across the 9 Binet A validation cohorts. The red line indicates the threshold selected as inclusion criteria in the final model.
Univariable and multivariable analysis for time to first treatment. (A) Forest plot of the hazard ratio for the 19 covariates assessed for association with TTFT by univariable analysis. Solid boxes indicate the hazard ratio, horizontal lines indicate the 95% CIs. The red dot line marks hazard ratio of 1. The bar graph on the left shows the multiplicity adjusted -log10 P value by Bonferroni. The red solid line marks the .05 significance level. (B) Forest plot of the hazard ratio for the 5 covariates assessed for association with TTFT by multivariable analysis. Solid boxes indicate the hazard ratio, horizontal lines indicate the 95% CIs. The red dot line marks hazard ratio of 1. The bar graph on the left shows the -log10 P value. The red solid line marks the .05 significance level. (C-D) The bar plot shows the percentage of covariate selection in the final multivariable model across the 9 Binet A validation cohorts. The red line indicates the threshold selected as inclusion criteria in the final model.
After pruning the model from the less consistent covariates, unmutated IGHV genes, absolute lymphocyte count higher than 15 × 109/L, and palpable lymph nodes were used to build the score and received a weight of 1 because of similar regression coefficients (Figure 1B). The hierarchical order of the sum of the scores in prognosticating TTFT was established by recursive partitioning analysis. This approach allowed us to establish the final international prognostic score to classify patients with asymptomatic CLL in early-stage disease (International Prognostic Score for Early-stage CLL [IPS-E]) according to the risk for treatment need. Patients with a score of 0 had the longest TTFT, and patients with a score of 2 or 3 had the shortest TTFT, whereas patients with a score of 1 had an intermediate TTFT (supplemental Figure 2).
Patients from the training cohort were segregated into 3 distinct risk categories according to the IPS-E (Figure 2A): low (score 0), intermediate (score 1), and high (score 2-3) risk. Sensitivity analysis accounting for death before treatment as competing risk consistently confirmed that IPS-E discriminates 3 risk groups with significantly different TTFT (supplemental Figure 3A). The ability of the IPS-E in discriminating TTFT (C-index) was 0.74 (Figure 3).
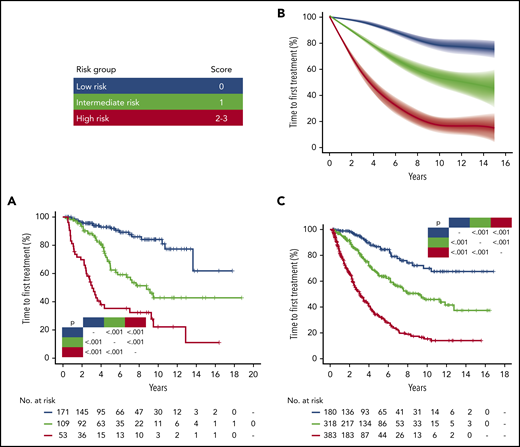
IPS-E stratified TTFT in patients with early-stage CLL managed with active surveillance. (A) Kaplan-Meier curves of TTFT stratified by IPS-E in the UEP discovery cohort. (B) Meta-analytic estimate of TTFT by IPS-E and the corresponding variability across the 9 Binet A validation cohorts. The bold line shows the cubic spline fitted on the meta-analytic estimate of the cumulative proportion of TTFT at each point. The shadow shows the cubic splines fitted on the meta-analytic estimate of the 95% CI of the cumulative proportion of TTFT at each point. (C) Kaplan-Meier curves of TTFT stratified by IPS-E in the Mayo Clinic validation cohort. Blue, low risk; green, intermediate risk; red, high risk by IPS-E. Multiplicity corrected P values by pairwise log-rank tests are shown.
IPS-E stratified TTFT in patients with early-stage CLL managed with active surveillance. (A) Kaplan-Meier curves of TTFT stratified by IPS-E in the UEP discovery cohort. (B) Meta-analytic estimate of TTFT by IPS-E and the corresponding variability across the 9 Binet A validation cohorts. The bold line shows the cubic spline fitted on the meta-analytic estimate of the cumulative proportion of TTFT at each point. The shadow shows the cubic splines fitted on the meta-analytic estimate of the 95% CI of the cumulative proportion of TTFT at each point. (C) Kaplan-Meier curves of TTFT stratified by IPS-E in the Mayo Clinic validation cohort. Blue, low risk; green, intermediate risk; red, high risk by IPS-E. Multiplicity corrected P values by pairwise log-rank tests are shown.
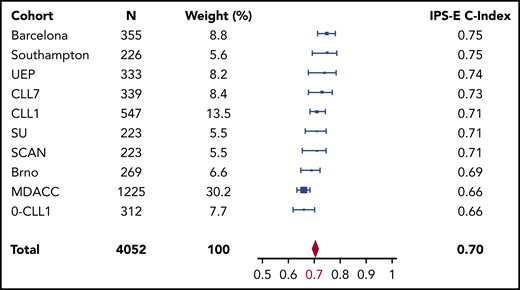
Meta-analysis of the estimate of the IPS-E discrimination capacity of TTFT across the study cohorts. Forest plot of the C-index of the 10 Binet A cohorts according to the IPS-E. Solid boxes indicate C-index in each study with dimensions proportional to sample size weights, horizontal lines indicate bootstrap 95% CIs. The red rhombus represents the overall estimate by the meta-analysis. MDACC, M.D. Anderson Cancer Center; SU, Sapienza University.
Meta-analysis of the estimate of the IPS-E discrimination capacity of TTFT across the study cohorts. Forest plot of the C-index of the 10 Binet A cohorts according to the IPS-E. Solid boxes indicate C-index in each study with dimensions proportional to sample size weights, horizontal lines indicate bootstrap 95% CIs. The red rhombus represents the overall estimate by the meta-analysis. MDACC, M.D. Anderson Cancer Center; SU, Sapienza University.
We validated the IPS-E, using 9 different independent series of patients with Binet stage A CLL. Some heterogeneity across the study cohorts allowed us to robustly confirm the external validity and generalizability of IPS-E irrespective of the case mix of the population. Heterogeneity in the baseline features reflected the source of the cohort; namely, population-based (SCAN: N = 223; median follow-up, 12.9 years; number of treatment events, 104) vs primary care institutions (Barcelona: N = 355; median follow-up, 10.3 years; number of treatment events, 155; Southampton: N = 226; median follow-up, 6.6 years; number of treatment events, 80; Sapienza University: N = 223; median follow-up, 11.3 years; number of treatment events, 117) vs referral centers (M.D. Anderson Cancer Center: N = 1225; median follow-up, 5.2 years; number of treatment events, 357; Brno: N = 269; median follow-up, 7.7 years; number of treatment events, 135) vs clinical trials (CLL1: N = 547; median follow-up, 8.3 years; number of treatment events, 229; CLL7: N = 339; median follow-up, 4.2 years; number of treatment events, 75; O-CLL-1: N = 312; median follow-up, 7.5 years; number of treatment events, 136). The IPS-E was confirmed and the 3 risk groups with significantly different TTFT were reproduced in the independent validation series both by primary analysis (Figure 2B; supplemental Figure 4A-I) and by sensitivity analysis, accounting for death before treatment as competing risk (supplemental Figure 3B-J). By meta-analysis across the cohorts, the IPS-E C-index for TTFT was 0.70 (Figure 3), whereas the C-index of the sole IGHV mutation status was 0.67 (95% CI, 0.66-0.68; supplemental Figure 5). Approaching the same meta-analytic assessment across the study cohorts, IPS-E showed a slightly higher discrimination capacity of TTFT compared with CLL-IPI (C-index, 0.68; 95% CI, 0.67-0.70; supplemental Figure 6).
The training and validation series were meta-analyzed to provide a more precise estimate of the risk for treatment need in each risk group (Figure 4). Low-risk patients overall corresponded to 29.5% of cases across the 10 Binet stage A CLL cohorts. Their cumulative risk for treatment need (not accounting for death resulting from competing risk) was less than 0.1% after 1 year of surveillance, and 8.4% after 5 years. The risk for treatment was 2.0 events per 100 person years among low-risk patients. Intermediate-risk patients overall corresponded to 36.2% of cases across the 10 Binet stage A CLL cohorts. Their cumulative risk for treatment need (not accounting for death resulting from competing risk) was 3.1% after 1 year of surveillance, and 28.4% after 5 years. The need for therapy was 6.1 events per 100 person years among intermediate-risk patients. High-risk patients overall corresponded to 34.3% of cases across the 10 validation cohorts. Their cumulative risk for treatment need (not accounting for death resulting from competing risk) was 14.1% after 1 year of surveillance and 61.2% after 5 years. The risk for treatment was 16.1 events per 100 person years among high-risk patients. IPS-E performed equivalently over sequential periods, which is consistent with the lack of updates of treatment initiation criteria in the guidelines3-5,21 and the steadiness of the threshold for commencement of therapy adopted over time by the physicians (supplemental Figure 7).
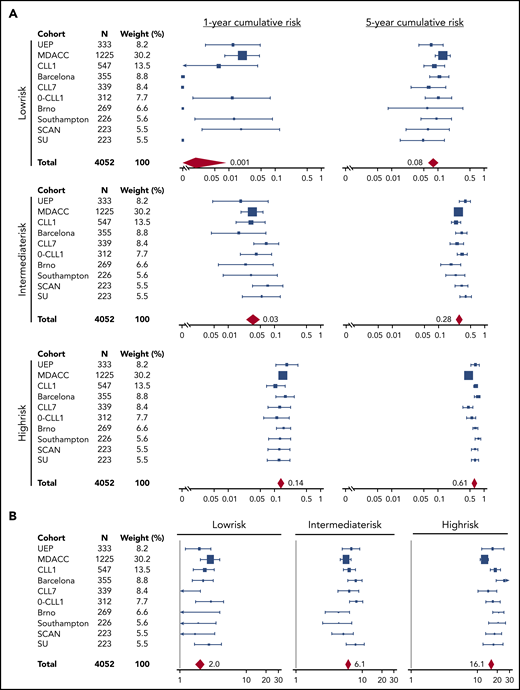
Meta-analysis of the estimates of the IPS-E cumulative incidence of treatment need across the study cohorts. Forest plots of the (A) 1- and 5-year cumulative incidence of treatment need across the 10 Binet A cohorts according to the IPS-E. (B) Forest plots of the number of treatment events per 100 person-years across the 10 Binet A cohorts according to the IPS-E. Solid boxes indicate the estimate in each study with dimensions proportional to sample size weights; horizontal lines indicate 95% CIs. The red rhombus represents the overall estimate by the meta-analysis.
Meta-analysis of the estimates of the IPS-E cumulative incidence of treatment need across the study cohorts. Forest plots of the (A) 1- and 5-year cumulative incidence of treatment need across the 10 Binet A cohorts according to the IPS-E. (B) Forest plots of the number of treatment events per 100 person-years across the 10 Binet A cohorts according to the IPS-E. Solid boxes indicate the estimate in each study with dimensions proportional to sample size weights; horizontal lines indicate 95% CIs. The red rhombus represents the overall estimate by the meta-analysis.
Two different clinical staging systems are used to stratify patients with CLL; namely, Binet staging, mostly used in European countries, and Rai staging, mostly used in the United States. We aimed at validating the IPS-E also in patients with early-stage CLL defined according to the Rai system (stage 0, I, and II) and managed with active surveillance (Mayo Clinic cohort: N = 881). The 3 IPS-E risk groups were well segregated both in the primary analysis (Figure 2C) and in the sensitivity analysis, accounting for death before treatment as competing risk (supplemental Figure 3K), thus confirming the generalizability of IPS-E also in patients with CLL staged according to the Rai system.
Discussion
The majority of patients with CLL are diagnosed in asymptomatic, early phases of the disease and are followed with no therapy.1,12 The clinical evolution of these patients is heterogeneous and difficult to predict. Although some patients display a quiescent disease never requiring therapy, others present active disease shortly after diagnosis and require intervention. Despite the general improvement on the outcome of patients with CLL, the relative survival of early-stage patients (ie, Binet stage A) has not significantly changed during the last decades.2 The management of these patients constitutes a major challenge and should be made on the basis of the best possible evidence and a risk-tailored policy.
There are several prognostic models that can be used to separate patients with different outcome within the whole population of subjects with CLL.14,18,19,35-40 However, in most of these models, the outcome of patients with early-stage CLL is either not analyzed nor investigated as a unique subgroup, and asymptomatic patients are not separately investigated. Moreover, in most such models, overall survival is the main end point.18,19 The uniqueness of patients with CLL diagnosed with asymptomatic early disease and the challenges posed by their management make advisable a specific prognostic model with TTFT as a main end point.
An important general caveat is that outcome indicators derived from patients requiring therapy do not necessarily apply to patients not requiring intervention. Among patients with CLL managed with active surveillance, the IGHV unmutated status is the biomarker with the strongest effect on TTFT prognostication. In contrast, other recurrent molecular features associated with inferior overall survival such as TP53 abnormalities, ATM abnormalities, and NOTCH1 mutations did not enter into the IPS-E model. Patients harboring TP53 aberrations, but mutated IGHV genes, may have a prolonged TTFT and a relatively benign clinical course under observation.41-43 Patients having ATM abnormalities or NOTCH1 mutations are highly enriched with unmutated IGHV genes. However, a large proportion of IGHV unmutated patients does not harbor ATM abnormalities or NOTCH1 mutation.41-46 Therefore, IGHV unmutated status not only captures ATM and NOTCH1 abnormalities but also expands its prognostic information to cases lacking these unfavorable genetic lesions.
The IPS-E presented in this study is a robust prognostic tool based on routine clinical and laboratory variables that informs at the time of diagnosis about the probability that a given patient with CLL in early-stage disease progresses and needs treatment. The cumulative risk for need of treatment after 1 and 5 years of observation was 14.1% and 61.2%, respectively, for an IPS-E high-risk patient, whereas it was 2.1% and 28.4%, for intermediate-risk patients and less than 0.1% and 8.4% for low-risk patients. Against this backdrop, IPS-E high-risk patients should be followed more closely than low- and intermediate-risk patients because of the likelihood of requiring treatment sooner.
IPS-E calculation is simple, being the sum of 3 variables. Compared with other scores, IPS-E calculation needs the assessment of only 1 molecular variable; namely, the IGHV mutation status, the testing of which is broadly available and standardized. IGHV status has been recognized by the current guidelines as a predictive biomarker for treatment tailoring.5,22,47 Our results further support the indication for testing IGHV mutations in patients with CLL. Moreover, as IGHV status never changes during the course of disease, it might be evaluated at the time of first diagnosis to provide an estimate of TTFT to the patient and to the treating physician. The results of our study also support that, as recommended by guidelines, FISH analysis and molecular testing for TP53 has no clinical utility when performed at CLL presentation in early-stage asymptomatic patients. TP53 abnormalities have a predictive role at time of therapy, but no prognostic role in an early-stage setting when determining TTFT. Also, they can change during the course of disease. Therefore, TP53 status may not be routinely evaluated in early-stage asymptomatic patients lacking treatment indication (in line with the iwCLL criteria and CLL guidelines).3-5,21 CLL can exhibit diverse growth patterns, including a continued stable state, never requiring therapy after diagnosis.48,49 However, a short lymphocyte doubling time is rarely an indication to start therapy by itself, as shown in previous reports50 and in this study, and therefore was not included as a variable for developing IPS-E.
The simplicity of IPS-E should facilitate its translation to the clinic. The IPS-E could help physicians, medical care providers, and health authorities in their strategies and resources allocation. Likewise, such a prognostic model could be useful to design clinical trials for patients with high-risk, early-stage CLL, an issue that has gained momentum because of the availability of effective small molecules with manageable toxicity.51,52
The development of the IPS-E followed an extensively used approach for the generation of prognostic scores in hematology.18,53-56 However, the risk for biases related to the timing of scheduled evaluations and premature censoring cannot be discarded. In contrast, although the IPS-E has an outcome discrimination capacity that is generally considered as acceptable57 and similar to that of other CLL prognostication scores now used in clinical practice and for clinical trial design,18 it can be eventually improved. Therefore, the IPS-E warrants prospective evaluation and can be regarded as a building block to which newly discovered independent outcome predictors for patients with early-stage CLL could be added.
Presented at the 15th International Conference on Malignant Lymphoma, Lugano, Switzerland, 18-22 June 2019.
Please e-mail the corresponding author for original data.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This study was supported by Swiss Cancer League, ID HSR-4660-11-2018, Bern, Switzerland; Research Advisory Board of the Ente Ospedaliero Cantonale, Bellinzona, Switzerland; European Research Council Consolidator Grant CLLCLONE ID: 772051; Grant No. 320030_169670/1 Swiss National Science Foundation, Berne, Switzerland; Fondazione Fidinam, Lugano, Switzerland; Nelia & Amadeo Barletta Foundation, Lausanne, Switzerland; Fond’Action, Lausanne, Switzerland; Translational Research Program, No. 6594-20, The Leukemia & Lymphoma Society, New York; AIRC 5x1000, No. 21198, Associazione Italiana per la Ricerca sul Cancro Foundation, Milan, Italy; PRIN 2017 No. 2015ZMRFEA_004, MIUR, Rome, Italy; Italian Ministry of Health 5x1000 funds 2014 and 2016, Compagnia S. Paolo Turin Italy, project 2017.0526; Swedish Cancer Society, Swedish Research Council, Knut and Alice Wallenberg Foundation, Karolinska Institutet, Karolinska University Hospital, and Radiumhemmets Forskningsfonder, Stockholm, Sweden; and MH CR grants No. NV19-03-00091 and DRO FNBr, 65269705, MEYS CR under the project CEITEC 2020 (LQ1601).
Authorship
Contribution: A.C. and L.T.d.B. performed statistical analysis, interpreted data, and wrote the manuscript; P.L., M. A. Hoechstetter, C.D.H., L.D.P., J.D., K.G.R., M. Gentile, M.D., F.R.M., G. Chiodin, M.M., J.B., J.K., K.E.S., R.M., M. A. Hess, and T.S.B. provided clinical data and contributed to manuscript revision; G. Cutrona, C.D., V.S., A.B., and A.N. contributed to molecular studies and manuscript revision; W.W., E.B., B.G., E.Z., S.G., M. Ghielmini, F.C., G.S., M.F., R.R., F.F., R.F., S.P., F.M., S.S., and H.D. contributed to manuscript revision; S.A.P., W.G.W., E.M., G.G., and M.H. provided key scientific insights and contributed to study design, data interpretation, and manuscript revision; and D.R. designed the study, interpreted data, and wrote the manuscript.
Conflict-of-interest disclosure: D.R. received honoraria from AbbVie, AstraZeneca, Gilead, Janssen, and Roche, and research grants from AbbVie, Gilead, Janssen, and Cellestia. M. Hallek received research support and honoraria from Roche, Gilead, Mundipharma, Janssen, Celgene, Pharmacyclics, and AbbVie. G.G. received honoraria from AbbVie, Janssen, AstraZeneca, and Sunesis. G.S. received honoraria from Celgene and Gilead. S.A.P. received research funding (to the institution) from Pharmacyclics, MorphoSys, Janssen, AstraZeneca, and Ascentage Pharma for clinical studies and honoraria from Pharmacyclics, AstraZeneca, Genentech, Gilead, and AbbVie (all to the institution). R.R. received honoraria from Illumina, Roche, Janssen, and AbbVie. R.F. received honoraria from AbbVie, Janssen, Amgen, Novartis, and AstraZeneca. F.F. received honoraria from Janssen-Cilag, and AbbVie, and a research grant from Gilead. C.D.H. received travel support and research funding from Roche. G. Cutrona received a research grant from Gilead (fellowship program 2017). The remaining authors declare no competing financial interests.
Correspondence: Davide Rossi, Department of Hematology, Oncology Institute of Southern Switzerland, Via Ospedale, 6500 Bellinzona, Switzerland; e-mail: davide.rossi@eoc.ch.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal