

TO THE EDITOR:
Burkitt lymphoma (BL) is a highly aggressive B-cell non-Hodgkin lymphoma (NHL),1 which accounts for 40% to 50% of pediatric NHL in nonendemic areas2 and ~80% of pediatric B-NHL in the developed world.3 The prognosis for patients with BL has improved dramatically in the past few decades with the use of intensive short courses of non-cross-resistant chemotherapy agents determined by the patient risk stratification.3 The prognosis has been further improved by the recent introduction of rituximab.4 Nevertheless, the prognosis for pediatric patients with relapsed/refractory (r/r) BL remains dismal, with an average overall survival rate of 25% or lower.5
Chimeric antigen receptor (CAR) T cells therapy for leukemia,6,7 and diffuse large B-cell lymphoma8,9 has shown promising efficacy and safety. However, data for CAR T-cell therapy in pediatric patients with BL are lacking.10,11 Here, we report the early response data of a small cohort of pediatric patients with r/r BL who were treated with CAR T-cell. This is a subcohort of patients from the clinical trial (ChiCTR1800014457) that we set up to study the efficacy and safety of CAR T-cell therapy for pediatric patients with mature B-cell lymphomas.
Five patients with r/r BL were enrolled, including 4 boys and 1 girl with a median age of 8 years (range, 6-10 years). Before enrollment in this trial, they all failed to show response to multiple courses of intensive chemotherapy and anti-CD20 monoclonal antibody treatment. Three patients were at stage IV and 2 were at stage III according to the St. Jude staging system. Other patient characteristics are listed in supplemental Table 1 on the Blood Web site. For CAR-T cell manufacturing, we routinely cryopreserve additional proportion of patients’ T cells in case a secondary or third round of CAR-T generation is needed. This is an unconventional approach, but was carried out for the following reasons: more technically feasible than attempting a multitargeted single CAR T-cell approach; presence of multiple targetable antigens on the patient's tumors; and the refractory progressive nature of the disease.
Patients 2, 4, and 5 (3/5) achieved complete remission (CR) after 1 round of CD19 CAR-T cell therapy (Figure 1A). Patient 1 showed transient response to CD19 CAR-T cell therapy. Because CD22 was also highly expressed on tumor cells (supplemental Table 2), we then treated the patient with CD22 CAR-T cell therapy as soon as the tumor regrowth was detected in the first course (Figure 1B). And the patient finally achieved CR. Patient 3 had no response to the first round of CD19 CAR-T treatment. Because the tumor cells expressed CD22 by immunohistochemical (IHC) stain analysis, we treated him with CD22 CAR-T. On day 45 postinfusion of CAR-T cells, 18F-fluorodeoxyglucose positron emission tomography-computed tomography revealed not only disappearance of the original tumor, but also the emergence of a new mass (supplemental Figure 1A-B). Based on the IHC reexamination of the new tumor cells (supplemental Table 2), we treated him with another round of CD20 CAR-T. On day 64, CR was confirmed by positron-emission tomography-computed tomography according to Cheson and colleagues.12
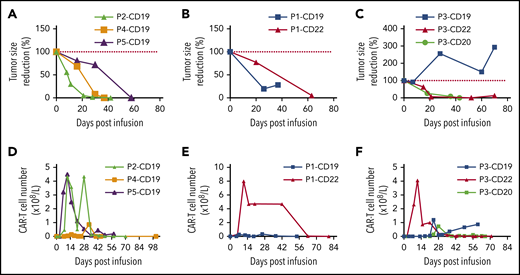
Tumor response and CAR-T cell expansion after infusion. (A) Tumor volume change (relative to tumor volume at day 0) for patients 2, 4, and 5. (B) Tumor volume change (relative to tumor volume at day 0) for patient 1 treated with CD19 and CD22 directed CAR-T cell therapy. (C) Tumor volume change (relative to tumor volume at day 0) for patient 3 treated with CD19, CD22, and CD20 directed CAR-T cell therapy. (D) CAR-T cells absolute number change for patients 2, 4, and 5. (E) CAR-T cells absolute number change for patient 1 treated with CD19 and CD22 directed CAR-T cell therapy. (F) CAR-T cells absolute number change for patient 3 treated with CD19, CD22, and CD20 directed CAR-T cell therapy. Single major tumor volume was measured by computed tomography or B-ultrasound.
Tumor response and CAR-T cell expansion after infusion. (A) Tumor volume change (relative to tumor volume at day 0) for patients 2, 4, and 5. (B) Tumor volume change (relative to tumor volume at day 0) for patient 1 treated with CD19 and CD22 directed CAR-T cell therapy. (C) Tumor volume change (relative to tumor volume at day 0) for patient 3 treated with CD19, CD22, and CD20 directed CAR-T cell therapy. (D) CAR-T cells absolute number change for patients 2, 4, and 5. (E) CAR-T cells absolute number change for patient 1 treated with CD19 and CD22 directed CAR-T cell therapy. (F) CAR-T cells absolute number change for patient 3 treated with CD19, CD22, and CD20 directed CAR-T cell therapy. Single major tumor volume was measured by computed tomography or B-ultrasound.
In summary, CR was achieved at days 64, 46, 64, 37, and 77 postinfusion for patients 1 to 5, respectively. The overall response rate of this multiple CAR-T infusion approach was 100%, including patients who did not respond or progressed after prior CAR-T products, with a median follow-up of 331 days, ranging from 149 to 428 days. Currently, all patients remain in CR. This high efficacy is consistent with results from previous study, which treated r/r B acute lymphoblastic leukemia using the same CAR-T cell products.13 And it is noteworthy that no cases in our study were bridged with hematopoietic stem cell transplantation after CAR-T cell therapy.
The in vivo expansion and persistence of CAR-T cells were monitored by flow cytometry (FC) (Figure 1D-F). After infusion, peak expansion of CAR-T cells in the blood occurred at about 2 weeks for patient 1 with CD22 CAR-T cells and for patients 2 and 5 with CD19 CAR-T cells. For patient 3 with CD20 CAR-T cells and patient 4 with CD19 CAR-T cells, CAR-T cells expansion peaked at about 4 and 5 weeks, respectively. The persistence of CAR-T cells also varied with a median duration of 42 days, ranging from 10 to 63 days. The seemingly inconsistency between the ongoing clinical response and FC-determined in vivo persistence of CAR-T cells is probably due to the relatively low sensitivity of FC. Low level of CAR-T cells persistence in the peripheral blood was confirmed by quantitative polymerase chain reaction for patients 3 and 4 during their recent follow-up (August 2019), with patient 3 showing 479 copies of CAR-vector DNA per microgram of genomic DNA and patient 4 showing 9 copies of CAR-vector DNA per microgram of genomic DNA. For patient 1, CD19 CAR-T cells failed to expand adequately (Figure 1E), which could be related to patient’s poor response. For patient 3, CD19 CAR-T cells expansion was significantly delayed, which might have caused the poor response (Figure 1F). The CD22 CAR-T cells seemed to expand adequately in patient 3, although not as robust as in patient 1 and in other previously reported patients with r/r B acute lymphoblastic leukemia,14 a new tumor was detected at day 45 postinfusion, however (Figure 1 F). After infusion, concentration of serum immunoglobulins G, A, and M were transiently decreased and maintained at subnormal level. Intravenous immunoglobulin was used to prevent potential infection.
Adverse events are summarized in supplemental Table 3. All patients showed myelosuppression, including anemia, thrombocytopenia, and neutropenia, likely attributed to lymphodepleting chemotherapy, which required blood transfusion. All patients showed cytokine release syndrome (CRS) of any grade.15 Four patients had hepatomegaly and elevated alanine transaminase without liver failure. Three patients had tachypnea, hypoxia, and pulmonary edema, which were controlled with oxygen inhalation and use of corticosteroids.
Two patients had infection, 1 of which was possible sepsis, the other was paronychia, which were managed with active supportive and antibiotics treatment. One patient developed hypotension and was treated with vasopressin.
All patients recovered fully after active symptomatic and supportive treatment including application of corticosteroids in CRS grade 3, but no patient received tocilizumab, which is the indicated therapy for CRS in the United States. The expedient use of corticosteroids alone to manage CRS is based on that corticosteroids may impair the function of T cells transiently, but may not affect objective and complete response rates nor the durability of responses,16 and importantly they have better accessibility in China and other developing countries. There were no death events. Serum levels of sCD25, interleukin-6 (IL-6), interferon-γ, and IL-10, which were reported to be the most significantly elevated cytokines,6 were monitored (supplemental Figure 3G-L). IL-10 is also a well-known anti-inflammation cytokine and sCD25 is reported to enhance T regulatory cell survival and function.17 The additional peak of IL-10 and sCD25 around day 50 in patient 1 indicated presence of a strong inhibitory immune response, which might contribute to the ineffectiveness of first round of therapy (supplemental Figure 3H,K).
In conclusion, CD19/CD20/CD22 CAR-T therapy showed promising efficacy for pediatric patients with r/r BL and the toxicities were tolerable. In addition, targeting different B-cell markers sequentially was effective for treating r/r patients, especially with our fast CAR-T cell manufacture technique, although the targeting sequence needs further optimization. A large cohort is needed to verify our findings and to address the concerns of the long-term efficacy of the sequential targeting approach in comparison with the multiantigen targeting approach.18
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their families for their understanding and participation and the staff of the Department of Laboratory at Beijing Boren Hospital for their technical assistance. The authors appreciate the careful reading and revision of the manuscript by Dong Chen (Division of Hematopathology, Mayo Clinic) and the critical discussion with Chunrong Tong and Jing Pan (Department of Hematology, Beijing Boren Hospital, Beijing).
Authorship
Contribution: W.Z. collected and analyzed the data and wrote the manuscript; Y.Z. designed and supervised the study, analyzed the data, and revised the manuscript; B.H., L.J., J.Y., J.D., S.W., and Y.L. collected the data and critically discussed the manuscript; C.Z. and Z.G. performed IHC and analyzed data; B.D. headed the CAR-T generation team; and A.H.C. provided CAR-T technique and support.
Conflict-of-interest disclosure: A.H.C. is a founding member of Shanghai YaKe Biotechnology Ltd., a biotechnology company focusing on research and development of tumor cellular immunotherapy. The remaining authors declare no competing financial interests.
Correspondence: Yonghong Zhang, Department of Pediatric Lymphoma, Beijing Boren Hospital; Department of Hematology Oncology, National Center for Children’s Health, Beijing Children’s Hospital, Capital Medical University, Beijing 100070, China; e-mail: zhangyongh@borenhospital.com.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal