

Key Points
RBC antibody–induced anemia is not a sufficient condition to predict a successful antibody for the treatment of a murine model of ITP.
RBC-specific antibodies that successfully block platelet phagocytosis in vitro predicted a successful outcome in a murine model of ITP.

Abstract
Polyclonal anti-D is a first-line therapy for immune thrombocytopenia (ITP). Monoclonal antibodies are desirable alternatives, but none have yet proven successful despite their ability to opsonize erythrocytes (or red blood cells, RBCs) and cause anemia. Here, we examined 12 murine erythrocyte–specific antibodies of different specificity and subtypes and found that 8 of these antibodies could induce anemia in antigen-positive mice. Of these 8 antibodies, only 5 ameliorated ITP. All antibodies were examined for their in vitro ability to support macrophage-mediated phagocytosis of erythrocytes. Antibodies which supported erythrocyte phagocytosis in vitro successfully ameliorated ITP in vivo. To examine the ability of each antibody to inhibit phagocytosis of platelets, the antibodies were used to sensitize erythrocytes in vitro and these were added to a platelet phagocytosis assay. Antibodies that inhibited platelet phagocytosis in vitro also all ameliorated ITP in vivo. We conclude that inducing anemia is not a sufficient condition for amelioration of ITP but that the antibody’s ability to prevent platelet phagocytosis in vitro predicted its ability to ameliorate ITP. We suggest that inhibition of in vitro platelet phagocytosis may prove to be a valuable tool for determining which erythrocyte antibodies would likely be candidates for clinical use in ITP.
Introduction
Anti-D is a donor-derived polyclonal immunoglobulin G (IgG) antibody directed against the RhD antigen on erythrocytes and can be used as a first-line treatment of immune thrombocytopenia (ITP).1,2 ITP is thought to be caused primarily (although not exclusively) due to platelet clearance by antiplatelet autoantibodies.1,2 It is generally accepted that antiplatelet antibodies, particularly against glycoprotein IIb/IIIa, mediate platelet destruction through the mononuclear phagocytic system (MPS).1-4
IVIg was first found to be efficacious in children with ITP.5 Salama then demonstrated that anti-D was also therapeutic.6 Although the mechanisms of both IVIg and anti-D remain to be elucidated, anti-D exerts its therapeutic effects at a 5 log–fold lower dose than IVIg.2
One of the proposed mechanisms, MPS blockade, posits that sensitized erythrocytes compete with the sensitized platelets for Fcγ receptor (FcγRs) sites in the MPS, particularly in the spleen.1,6 This hypothesis is supported by studies where anti-D was inefficacious in ITP patients lacking the RhD antigen.7,8 Interestingly, despite the ability of anti-D to lower hemoglobin values in some patients, no relationship is apparent between hemoglobin decreases and responses in ITP9-11 or between erythrocyte sensitization by different doses of anti-D and responses in ITP.9,10
To add to the complexity, we have recently shown that the TER119 antibody, which mimics the action of anti-D in murine ITP amelioration,12-14 has anti-inflammatory effects in murine arthritis and can prevent disease activity in a murine model of transfusion-related acute lung injury.15 Since the spleen and the MPS are not a priori involved in the development of arthritis or transfusion-related acute lung injury, this could question the simplistic concept of MPS blockade in the therapeutic action of anti-erythrocyte antibodies.
To the best of our knowledge, all potential anti-D replacement antibodies tested to date have led to disappointing results. Better knowledge of its mechanism is required to move forward.
Methods
Details are provided in supplemental Methods (available on the Blood Web site).
Results and discussion
The mechanism of action of anti-D in ITP amelioration remains unclear. In this study, 1 polyclonal and 11 monoclonal anti-erythrocyte antibodies were analyzed to determine the features characteristic of therapeutic antibodies in passive murine ITP. The specificity, IgG subtype, ability to sensitize erythrocytes, induce anemia, mediate erythrocyte phagocytosis, inhibit platelet phagocytosis, and ameliorate ITP were evaluated. The data are summarized in Table 1 with detailed results of the prototype antibody (TER119, rat IgG2b) in Figure 1. Data for the remaining antibodies are provided in supplemental Figures 1-4.
Characteristics and results summary of the anti-erythrocyte antibodies studied
| Antibody | Isotype | Antigen | Relative binding to erythrocytes* | Anemia† | Induces erythrocyte phagocytosis | Inhibits platelet phagocytosis | Ameliorates ITP |
|---|---|---|---|---|---|---|---|
| TER119 | Rat IgG2b | mGPA | ++++ | ++++ | Yes | Yes | Yes |
| Deglycosylated TER119 | Rat IgG2b | mGPA | ++++ | ++++‡ | No | No | No |
| TER119 | Mouse IgG1 | mGPA | ++++ | ++++ | Yes | Yes | Yes |
| TER119 | Mouse IgG2a | mGPA | ++++ | ++++ | Yes | Yes | Yes |
| M1/69 | Rat IgG2b | CD24 | ++ | +++§ | Yes | Yes | Yes |
| 34-3C | Mouse IgG2a | Band3 | ++++ | +++ | Yes | Yes | Yes |
| 4B7 | Mouse IgG1 | HEL | ++ | − | No | No | No |
| 5B9 | Mouse IgG1 | HEL | ++ | − | No | No | No |
| MIMA29 | Mouse IgG2a | Duffy | ++++ | +++ | No | No | No |
| CBC512 | Mouse IgG1 | Duffy | +++ | ++++ | No | No | No |
| Anti-OVA | Rabbit polyclonal | OVA | ++ | −|| | No | No | No |
| 6A7 | Mouse IgG1 | hGPA | ++ | −|| | No | N/T | No |
| Antibody | Isotype | Antigen | Relative binding to erythrocytes* | Anemia† | Induces erythrocyte phagocytosis | Inhibits platelet phagocytosis | Ameliorates ITP |
|---|---|---|---|---|---|---|---|
| TER119 | Rat IgG2b | mGPA | ++++ | ++++ | Yes | Yes | Yes |
| Deglycosylated TER119 | Rat IgG2b | mGPA | ++++ | ++++‡ | No | No | No |
| TER119 | Mouse IgG1 | mGPA | ++++ | ++++ | Yes | Yes | Yes |
| TER119 | Mouse IgG2a | mGPA | ++++ | ++++ | Yes | Yes | Yes |
| M1/69 | Rat IgG2b | CD24 | ++ | +++§ | Yes | Yes | Yes |
| 34-3C | Mouse IgG2a | Band3 | ++++ | +++ | Yes | Yes | Yes |
| 4B7 | Mouse IgG1 | HEL | ++ | − | No | No | No |
| 5B9 | Mouse IgG1 | HEL | ++ | − | No | No | No |
| MIMA29 | Mouse IgG2a | Duffy | ++++ | +++ | No | No | No |
| CBC512 | Mouse IgG1 | Duffy | +++ | ++++ | No | No | No |
| Anti-OVA | Rabbit polyclonal | OVA | ++ | −|| | No | No | No |
| 6A7 | Mouse IgG1 | hGPA | ++ | −|| | No | N/T | No |
HEL, hen egg lysosome; hGPA, human glycophorin A protein; N/T, not tested; mGPA, mouse glycophorin A–associated protein; OVA, ovalbumin.
Antibody binding to erythrocytes was quantified based on the median fluorescence intensity (MFI) at the saturating and/or the highest concentration tested (–≤50, + ≤100, ++ ≤250, +++ ≤500, ++++ ≥500).
Anemia refers to the ability of the antibody to leading to reductions in erythrocyte concentration in circulation (−, <6.25%; +, >6.25%; ++, >12.5%; +++, >25%; ++++, >50% reduction).
TER119 is able to cause anemia by an FcγR,13 macrophage-mediated16 pathway as well as by a cation-driven FcγR/complement-independent pathway.17 As deglycosylation of TER119 would impair FcγR/complement-dependent events, the anemia observed by the deglycosylated TER119 would be anticipated to occur due to the cation-driven FcγR/C3-independent pathway.
The degree of anemia induced by M1/69 varied across the mice strains tested (+++ for C57Bl/6, ++ for BALB/c, and − for CD-1).
Anti-OVA– and 6A7-induced anemia was recorded daily for 6 and 2 days only, respectively (as opposed to daily reading over 8 days).
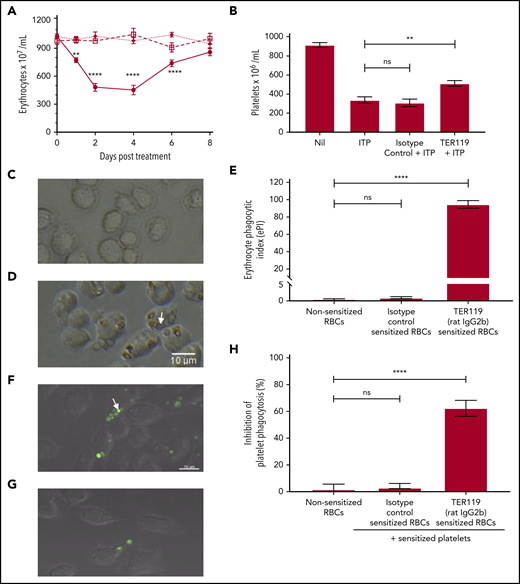
Characterization of TER119 (rat IgG2b) in vitro and in vivo. (A) To assess anemia, mice were uninjected (□—□), injected with 40 µg isotype control antibody (◆—◆), or injected with 40 µg TER119 antibody (●–●). Mice were bled and erythrocytes enumerated at the indicated time points. Day 0 counts represent the erythrocyte count before treatment; n = 5-8. Statistical significance was determined using a 2-way analysis of variance followed by Tukey’s test. (B) To test for therapeutic efficacy in ITP, mice were injected with the antierythrocyte antibody followed by MWReg30 (antiplatelet antibody) after 30 minutes to induce thrombocytopenia. Platelet counts were assessed 24 hours later. “Nil” represents the baseline platelet count of the control untreated mice, while ITP refers to the mice receiving the antiplatelet antibody alone (with no therapeutic treatment); n = 6-7. Statistical significance was determined using the Mann-Whitney U test. (C-E) Erythrocyte phagocytosis. Erythrocytes were sensitized with the isotype control antibody (C) or TER119 antibody (D) and incubated with RAW 264.7 macrophages to assess phagocytosis. Phagocytosis was observed using an inverted light microscope. Engulfed erythrocytes (D; arrow points to an engulfed erythrocyte) are seen as a red-brown sphere within macrophages. Phagocytosis was quantified using the erythrocyte phagocytic index (ePI; E); n = 4-8. Statistical significance was determined using the Mann-Whitney U test. (F-H) Inhibition of platelet phagocytosis. Platelets were labeled with a cell tracker dye (green) and sensitized with MWReg30 (antiplatelet antibody). Erythrocytes were sensitized with the isotype control antibody (F) or TER119 antibody (G). Sensitized platelets and erythrocytes were simultaneously incubated with RAW 264.7 macrophages. Platelet phagocytosis was assessed by confocal microscopy. Engulfed platelets (F; arrow points to an engulfed platelet) are seen as a green sphere within macrophages. Inhibition of platelet phagocytosis (H) was calculated relative to the sensitized platelets group for that experiment (no treatment; arbitrarily set as 0% inhibition); n = 3-6. Statistical significance was determined using the Mann-Whitney U test. ** P < .01, **** P < .0001 (ns, not significant).
Characterization of TER119 (rat IgG2b) in vitro and in vivo. (A) To assess anemia, mice were uninjected (□—□), injected with 40 µg isotype control antibody (◆—◆), or injected with 40 µg TER119 antibody (●–●). Mice were bled and erythrocytes enumerated at the indicated time points. Day 0 counts represent the erythrocyte count before treatment; n = 5-8. Statistical significance was determined using a 2-way analysis of variance followed by Tukey’s test. (B) To test for therapeutic efficacy in ITP, mice were injected with the antierythrocyte antibody followed by MWReg30 (antiplatelet antibody) after 30 minutes to induce thrombocytopenia. Platelet counts were assessed 24 hours later. “Nil” represents the baseline platelet count of the control untreated mice, while ITP refers to the mice receiving the antiplatelet antibody alone (with no therapeutic treatment); n = 6-7. Statistical significance was determined using the Mann-Whitney U test. (C-E) Erythrocyte phagocytosis. Erythrocytes were sensitized with the isotype control antibody (C) or TER119 antibody (D) and incubated with RAW 264.7 macrophages to assess phagocytosis. Phagocytosis was observed using an inverted light microscope. Engulfed erythrocytes (D; arrow points to an engulfed erythrocyte) are seen as a red-brown sphere within macrophages. Phagocytosis was quantified using the erythrocyte phagocytic index (ePI; E); n = 4-8. Statistical significance was determined using the Mann-Whitney U test. (F-H) Inhibition of platelet phagocytosis. Platelets were labeled with a cell tracker dye (green) and sensitized with MWReg30 (antiplatelet antibody). Erythrocytes were sensitized with the isotype control antibody (F) or TER119 antibody (G). Sensitized platelets and erythrocytes were simultaneously incubated with RAW 264.7 macrophages. Platelet phagocytosis was assessed by confocal microscopy. Engulfed platelets (F; arrow points to an engulfed platelet) are seen as a green sphere within macrophages. Inhibition of platelet phagocytosis (H) was calculated relative to the sensitized platelets group for that experiment (no treatment; arbitrarily set as 0% inhibition); n = 3-6. Statistical significance was determined using the Mann-Whitney U test. ** P < .01, **** P < .0001 (ns, not significant).
All antibody preparations bound erythrocytes, but only 8 induced anemia. With some exceptions, antibodies leading to higher erythrocyte sensitization generally induced more anemia (Table 1). More dramatic was the observation that antibodies that caused anemia did not necessarily ameliorate ITP (supplemental Figure 1). In particular, all forms of TER119 (rat IgG2b, deglycosylated rat IgG2b, mouse IgG1, and mouse IgG2a), 34-3C, MIMA29, and CBC512 induced significant anemia, yet only some ameliorated ITP (Table 1,13,16,17 columns 5 vs 8). Interestingly, M1/69 ameliorated ITP and induced a more moderate level of anemia and was therefore explored in additional strains of mice. M1/69 ameliorated ITP in C57BL/6, BALB/c, and CD-1 mice (supplemental Figure 2A) but only induced anemia in C57BL/6 and BALB/c strains (supplemental Figure 2B). ITP amelioration appeared independent of complement component 3 (C3); TER119 (IgG2a) and M1/69 were tested in C3−/− mice, and increased platelet counts were comparable to C57BL/6 mice (not shown).
Three therapeutically successful variants of TER119 (rat IgG2b, mouse IgG1, and mouse IgG2a) and 1 unsuccessful variant (deglycosylated rat IgG2b) were also evaluated in the ITP treatment model, where thrombocytopenia is induced first by daily injections of the antiplatelet antibody and the erythrocyte antibodies are then used as a therapeutic.14 In this therapeutic model, the results mirrored those in the classic passive ITP model (supplemental Figure 5).
Contrasting research has argued for and against the role of anemia in ITP amelioration by anti-D.6,9,10,18-20 In the only clinical study testing a single monoclonal anti-D in ITP, all 7 patients showed signs of hemolysis and/or anemia, while only 1 patient minimally responded, thus implying that anemia is not sufficient to ameliorate ITP.21 This conclusion is also supported by studies evaluating polyclonal anti-D, where responses did not correlate with erythrocyte-sensitization, hemolysis, hemoglobin, or haptoglobin levels.6,9,10,18,19,22
We have previously shown that TER119 ameliorates ITP before signs of anemia and does not have a therapeutic effect concordant with maximal anemia,15 indicating that anemia itself may be uncoupled from therapeutic activity. In the present work, 3 antibodies (deglycosylated TER119, CBC512, and MIMA29) induced anemia but failed to ameliorate murine ITP, suggesting that anemia itself is insufficient to ameliorate ITP by anti-erythrocyte antibodies.
These results indicate no absolute requirement of a particular murine subtype and its therapeutic activity in ITP. At least 1 antibody from all IgG subclasses tested (IgG1, IgG2a, and IgG2b) ameliorated ITP. However, a deglycosylated variant of TER119 did not ameliorate ITP, indicating the importance of a functional Fc region. ITP amelioration was also independent of the antigen specificity; 3 antibodies, directed against 3 different antigens, were therapeutic, suggesting that targets other than the RhD antigen can be explored when developing anti-D replacements.
Assessing the ability of each antibody to induce phagocytosis, only antibodies that ameliorated ITP supported erythrocyte phagocytosis (Table 1; column 6 vs 8). Going a step further, it was observed that platelet phagocytosis was greatly diminished upon addition of erythrocytes sensitized with (only) the phagocytosis-inducing antibodies (Table 1; column 6 vs 7). These in vitro results were generated in a complement-depleted system (heat-inactivated serum, erythrocytes were collected in a citrate buffer and plasma removed prior to use).
Rat IgG2b and mouse IgG2a (similar to human IgG3 in ability to engage multiple FcγRs23 ) were superior to murine IgG1 antibodies in phagocytosis. Human anti-D of the IgG3 subtype is also more effective than IgG1 at inhibiting platelet phagocytosis.24 Previous monoclonal anti-D replacements for ITP have been of the IgG1 subtype.21,25 It is suggested that a human IgG3 antibody (potentially re-engineered to extend half-life) may be worth considering.
Rozrolimupab (cocktail of 25 monoclonal anti-D antibodies) proved efficacious, with a response rate of 62%; however, a dose four- to sixfold higher than anti-D was used, and 75% of patients experienced adverse events.2,25 The individual profiles of each of the antibodies in Rozrolimupab are not known. While it is certainly possible that some (or all) of the antibodies in Rozrolimupab conferred therapeutic activity and caused toxicity, it is also possible that some antibodies were therapeutic, some led to adverse events and some were without effect.
In the current work, we show that only antibodies that induced erythrocyte phagocytosis and prevented platelet phagocytosis in vitro were successful in preventing thrombocytopenia. It should be noted that while our murine and in vitro studies can be used to model the clinical setting, they cannot adequately explore all the facets of the human pathophysiology involved. In addition, although our results suggest that complement is not a crucial factor here, we cannot completely rule out its involvement. Our findings suggest that the ability of an anti-erythrocyte antibody to ameliorate ITP may not be directly related to the subtype of the antibody, antigen specificity, extent of erythrocyte sensitization, or anemia. Rather, its therapeutic activity in ITP can be predicted by assessing in vitro phagocytosis. This relationship may prove useful when developing and testing monoclonal replacements for anti-D.
Requests for data sharing should be e-mailed to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank James C. Zimring for providing the transgenic HOD mouse model and HEL-specific antibodies (4B7, 5B9) and Jeanne Hendrickson for the hGPA antibody (6A7). The authors also thank Marion E. Reid and Gregory Halverson for their donation of MIMA29, Makoto Uchikawa for CBC512, and Mohandas Narla for the transgenic hGPA mouse model. The authors are also thankful to Jennifer Brasseit, Ian K. Campbell, Fabian Käsermann, Sandra Koernig, and Adrian W. Zuercher from CSL Behring for providing the IgG1 and IgG2a variants of TER119. The authors also thank colleagues Andrew Crow, Joan Legarda, Yoelys Cruz-Leal, Yawen Wang, Gurleen Kaur, Hanna Wabnitz, Yuexin Shan, Danielle Marjoram, and the St. Michael’s Hospital Research Vivarium and the Core Facilities staff. In particular, they thank Caterina Di Ciano-Oliveria for her outstanding help and assistance with the bioimaging studies.
This work was supported by Canadian Blood Services (intramural grant), funded by the federal government (Health Canada) and the provincial and territorial ministries of health.
The views expressed herein do not necessarily represent the views of Canadian Blood Services or the federal, provincial, or territorial governments of Canada.
Authorship
Contribution: R.K. designed research, performed experiments, analyzed data, and wrote the manuscript; C.-C.J. and M.M. designed research, performed experiments, and analyzed data; X.C. and P.A.A.N. performed experiments and analyzed data; and A.H.L. designed research, analyzed data, obtained grant funding, and wrote the manuscript.
Conflict-of-interest disclosure: A.H.L. has patents on the use of monoclonal antibodies to treat ITP and has received research funding from CSL Behring and Rigel Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Alan H. Lazarus, 209 Victoria St, Room 422, Keenan Research Centre in LKSKI, St. Michael’s Hospital, Toronto, ON M5B 1T8, Canada; e-mail: alan.lazarus@unityhealth.to.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal