


Abstract
The nucleophosmin (NPM1) gene encodes for a multifunctional protein with prominent nucleolar localization that shuttles between nucleus and cytoplasm. NPM1 mutations represent the most common genetic lesion in adult acute myeloid leukemia (AML; about one third of cases), and they act deterministically to cause the aberrant cytoplasmic delocalization of NPM1 mutants. Because of its unique features, NPM1-mutated AML is recognized as a distinct entity in the 2017 World Health Organization (WHO) classification of hematopoietic neoplasms. Here, we focus on recently identified functions of wild-type NPM1 in the nucleolus and address new biological and clinical issues related to NPM1-mutated AML. The relevance of the cooperation between NPM1 and other mutations in driving AML with different outcomes is presented. We also discuss the importance of eradicating NPM1-mutated clones to achieve AML cure and the impact of preleukemic clonal hematopoiesis persistence in predisposing to second AML. The contribution of HOX genes’ expression to the development of NPM1-mutated AML is also highlighted. Clinically, yet unsolved diagnostic issues in the 2017 WHO classification of myeloid neoplasms and the importance of NPM1 mutations in defining the framework of European LeukemiaNet genetic-based risk stratification are discussed. Finally, we address the value and limits of NPM1-based measurable residual disease assessment for treatment guidance and present the results of promising preclinical studies with XPO1 and menin-MLL inhibitors.
Introduction
The nucleophosmin (NPM1) gene encodes for a ubiquitous multifunctional shuttling protein1 with predominant nucleolar localization. NPM1 is the most commonly mutated gene in adult acute myeloid leukemia (AML; ∼30% of cases).2 The most distinguishing feature of NPM1 mutants is their aberrant cytoplasmic localization,3 which led to the discovery of NPM1 mutations by immunohistochemistry (IHC), prior to the next-generation sequencing (NGS) era.2 It was a long journey before NPM1-mutated AML was recognized as a distinct entity in the 2017 World Health Organization (WHO) classification of hematopoietic neoplasms.4,5 Together with double-CEBPA–mutated AML, NPM1-mutated AML is 1 of the 2 WHO leukemia entities defined by a single-gene mutation. Here, we focus on new insights about NPM1 wild-type (NPM1wt) protein and NPM1-mutated AML.
NPM1: a multifunctional nucleolar protein with shuttling properties
NPM1wt is a nucleolar chaperone protein, active as an oligomer (pentamer/decamer). The structure and putative functions of NPM1wt have been reviewed extensively6-9 and are summarized in Figure 1. Here, we focus on newly described nucleolar functions of NPM1wt.

Structure and mutliple functions of NPM1wt. (A) Structure of NPM1wt and mutant NPM1 (NPM1mut) for comparison. NPM1wt and NPM1mut share the same structure, only differing in the very last portion of the C terminus. The hydrophobic N terminus contains 2 weak leucine-rich nuclear export signals (NESs) and a self-oligomerization core responsible for the formation of NPM1 pentamers. The N terminus also acts as a chaperone by preventing protein misfolding in the nucleolus. The central portion of NPM1wt is highly disordered and acidic because it contains two 10- to 20-aa stretches of aspartic and glutamic acid residues whose negative charge is involved in binding of NPM1wt to histones. The central region also contains a bipartite NLS driving the protein from the cytoplasm to the nucleoplasm. The C-terminal basic domain of NPM1wt mediates binding to nucleic acids and TP53. The very last portion of the NPM1wt C terminus, with its highly conserved W288 and W290, forms a globular structure consisting of a 3-helix bundle, which is responsible for its nucleolar localization signal (NoLS). Compared with NPM1wt, the C terminus of NPM1mut loses W288 and W290 (or W290 alone) and gains a new NES. (B) Summary of the multiple functions of NPM1wt. NPM1wt inhibits centrosome duplication (1). During the cell cycle, NPM1wt is released from the centrosome to allow its duplication and the formation of the mitotic spindle. With its predominant nucleolar localization and the ability to bind rRNA and ribosomal proteins, NPM1wt cooperates in ribosome biogenesis (2). NPM1wt is also a histone chaperone (3) with the ability to bind core and linker histones and to regulate the formation of perinucleolar heterochromatin. It is also involved in DNA repair (4); NPM1wt binds to TP53, enhancing its stability and transcriptional activity, as well as to APE1, regulating its endonuclease activity depending on the type of DNA damage. All of the above functions have been reviewed extensively.6-9 More details regarding the involvement of NPM1wt in other recently reported functions, such as 2′-O-methylation of rRNA (5) and LLPS in the nucleolus (6) are provided in the text. (C) Schematic representation of NPM1wt behavior upon nucleolar stress. Nucleolar stress through the inhibition of RNA polymerase I results in the spread of NPM1wt from the nucleolus to the nucleoplasm. There, NPM1wt can bind and inhibit HDM2, a protein with E3 ligase activity that promotes TP53 degradation through the proteasome. HDM2 inhibition results in increased levels of TP53, cell cycle arrest, and apoptosis. FBL, fibrillarin; NIPBL, nipped-B-like protein; snoRNA, small nucleolar RNA.
Structure and mutliple functions of NPM1wt. (A) Structure of NPM1wt and mutant NPM1 (NPM1mut) for comparison. NPM1wt and NPM1mut share the same structure, only differing in the very last portion of the C terminus. The hydrophobic N terminus contains 2 weak leucine-rich nuclear export signals (NESs) and a self-oligomerization core responsible for the formation of NPM1 pentamers. The N terminus also acts as a chaperone by preventing protein misfolding in the nucleolus. The central portion of NPM1wt is highly disordered and acidic because it contains two 10- to 20-aa stretches of aspartic and glutamic acid residues whose negative charge is involved in binding of NPM1wt to histones. The central region also contains a bipartite NLS driving the protein from the cytoplasm to the nucleoplasm. The C-terminal basic domain of NPM1wt mediates binding to nucleic acids and TP53. The very last portion of the NPM1wt C terminus, with its highly conserved W288 and W290, forms a globular structure consisting of a 3-helix bundle, which is responsible for its nucleolar localization signal (NoLS). Compared with NPM1wt, the C terminus of NPM1mut loses W288 and W290 (or W290 alone) and gains a new NES. (B) Summary of the multiple functions of NPM1wt. NPM1wt inhibits centrosome duplication (1). During the cell cycle, NPM1wt is released from the centrosome to allow its duplication and the formation of the mitotic spindle. With its predominant nucleolar localization and the ability to bind rRNA and ribosomal proteins, NPM1wt cooperates in ribosome biogenesis (2). NPM1wt is also a histone chaperone (3) with the ability to bind core and linker histones and to regulate the formation of perinucleolar heterochromatin. It is also involved in DNA repair (4); NPM1wt binds to TP53, enhancing its stability and transcriptional activity, as well as to APE1, regulating its endonuclease activity depending on the type of DNA damage. All of the above functions have been reviewed extensively.6-9 More details regarding the involvement of NPM1wt in other recently reported functions, such as 2′-O-methylation of rRNA (5) and LLPS in the nucleolus (6) are provided in the text. (C) Schematic representation of NPM1wt behavior upon nucleolar stress. Nucleolar stress through the inhibition of RNA polymerase I results in the spread of NPM1wt from the nucleolus to the nucleoplasm. There, NPM1wt can bind and inhibit HDM2, a protein with E3 ligase activity that promotes TP53 degradation through the proteasome. HDM2 inhibition results in increased levels of TP53, cell cycle arrest, and apoptosis. FBL, fibrillarin; NIPBL, nipped-B-like protein; snoRNA, small nucleolar RNA.
New functions of NPM1wt in the nucleolus
The nucleolus, a membrane-less organelle, is formed through a “liquid-liquid” phase separation (LLPS) process10-12 that causes the segregation of NPM1wt and other nucleolar components from the surrounding nucleoplasm. This process is similar to how oil and water separate from each other when mixed. LLPS is facilitated by the interaction between pentameric NPM1wt, proteins containing R-motifs (ie, multivalent arginine-rich linear motifs [sharing features with nucleolar localization signals (NoLSs)] and nascent ribosomal RNA [rRNA]).10,13 These heterotypic interactions are critical for nucleolar phase separation and for maintaining the directionality of ribosome biogenesis through the different subcellular compartments (ie, from the phase-separated nucleolus to the nonphase-separated nucleoplasm).13 Examples include the interactions of NPM1wt with p14Arf R-motifs14 or with SURF6 and rRNA.13
The nucleolus is made-up of a fibrillar core, surrounded by the dense fibrillar component and the granular component (GC). The multilayered structure of the nucleolus is determined by LLPS between nucleolar proteins, primarily NPM1wt and fibrillarin that segregate in the GC and dense fibrillar component, respectively11 (Figure 1B). Within the GC phase, NPM1wt serves as a scaffolding protein for nascent rRNA13 and helps prevent irreversible aggregation of cytoplasmic misfolded proteins migrating to the nucleolus following stress.15
NPM1wt also participates in rRNA 2′-O-methylation16 (Figure 1B). Specifically, NPM1wt binds directly to several C/D box small nucleolar RNAs16 and to the methyltransferase fibrillarin. The fibrillarin–NPM1–small nucleolar RNA complex actively methylates rRNA, and NPM1wt loss results in altered rRNA 2′-O-methylation and deregulated translation.16 Accordingly, germline loss-of-function mutations in the NPM1 acidic region may contribute to the pathogenesis of dyskeratosis congenita,16 a well-known ribosomopathy. However, the role of altered translation in NPM1-mutated AML remains to be established.
Intracellular shuttling of NPM1wt
The nucleus-cytoplasm shuttling of NPM1wt is regulated by specific signal motifs3 (Figures 1A and 2A). A bipartite nuclear localization signal (NLS), recognized by importin α/β,17 drives NPM1wt from the cytoplasm to the nucleoplasm. NPM1wt then localizes to the nucleolus through an aromatic NoLS, a 3-helix structure located at the C terminus.18,19 NoLS contains 2 tryptophans (W288 and W290) that stabilize the hydrophobic core of triple helix, enabling its proper folding. This is critical for localizing NPM1wt to the nucleolus18 through interaction with G-quadruplexes of ribosomal DNA.20
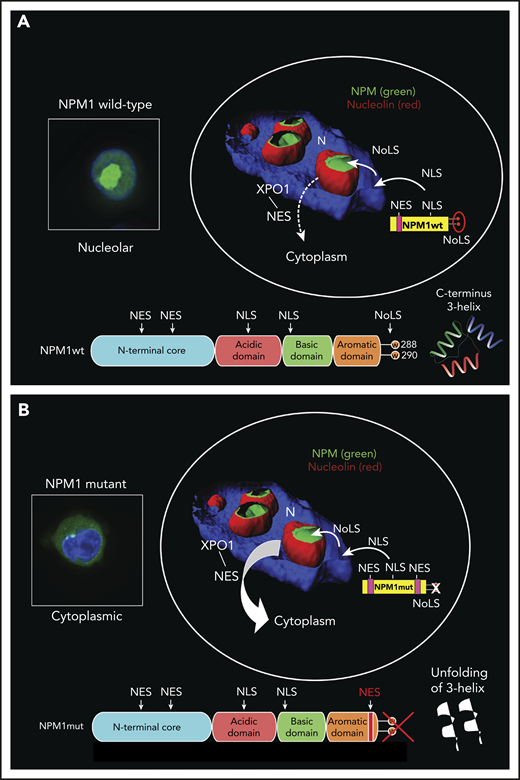
Nucleo-cytoplasmic shuttling of NPM1wt and NPM1 mutants. (A) Physiological NPM1wt shuttling. The bipartite NLS drives the nuclear import of NPM1wt. The C-terminal 3-helix NoLS dictates NPM1wt positioning within the nucleolus, whereas the 2 weak nuclear export signals (NESs) at the N terminus are responsible for its export from the nucleus to the cytoplasm (dashed arrow). Eventually, the nuclear import predominates over export so that almost all NPM1wt resides in the nucleolus. (B) Abnormal traffic of NPM1 mutants. Changes in tryptophans (W) 288 and 290 (or 290 alone), with consequent unfolding of the C-terminal triple-helix, abrogate the capability of the mutant to reside within the nucleolus. Furthermore, the insertion of a third NES motif at the C terminus markedly increases the export of the protein (thick arrow). Thus, the nuclear export predominates over the import, and the NPM1 mutant is delocalized in the cytoplasm. The 3-dimensional reconstruction of confocal images shown in the circles has been adapted from Falini et al.7 Nuclei are stained with 4′,6-diamidino-2-phenylindole, while the nucleoli are double-stained for NPM1 and nucleolin. Boxes show OCI-AML3 cells engineered to express either endogenous NPM1wt or NPM1 mutant fused to GFP. Nuclei are stained with Hoechst 33342.
Nucleo-cytoplasmic shuttling of NPM1wt and NPM1 mutants. (A) Physiological NPM1wt shuttling. The bipartite NLS drives the nuclear import of NPM1wt. The C-terminal 3-helix NoLS dictates NPM1wt positioning within the nucleolus, whereas the 2 weak nuclear export signals (NESs) at the N terminus are responsible for its export from the nucleus to the cytoplasm (dashed arrow). Eventually, the nuclear import predominates over export so that almost all NPM1wt resides in the nucleolus. (B) Abnormal traffic of NPM1 mutants. Changes in tryptophans (W) 288 and 290 (or 290 alone), with consequent unfolding of the C-terminal triple-helix, abrogate the capability of the mutant to reside within the nucleolus. Furthermore, the insertion of a third NES motif at the C terminus markedly increases the export of the protein (thick arrow). Thus, the nuclear export predominates over the import, and the NPM1 mutant is delocalized in the cytoplasm. The 3-dimensional reconstruction of confocal images shown in the circles has been adapted from Falini et al.7 Nuclei are stained with 4′,6-diamidino-2-phenylindole, while the nucleoli are double-stained for NPM1 and nucleolin. Boxes show OCI-AML3 cells engineered to express either endogenous NPM1wt or NPM1 mutant fused to GFP. Nuclei are stained with Hoechst 33342.
The nuclear export of NPM1wt is mediated by the interaction of 2 N-terminal nuclear export signals (NESs) with the nuclear exporter XPO1 (also named CRM1).7 Binding of XPO1 to some of its cargo occurs within the nucleolus.21 We hypothesize that localization of NPM1wt to the GC, the external component of the nucleolus,11 may facilitate the access of XPO1 to this area and its interaction with NPM1wt. However, NPM1wt is not exported efficiently, because the N-terminal NESs have low affinity for XPO1.17 We believe that nuclear export may also be weak because it is opposed by multiple forces that anchor NPM1wt to the nucleolus, including binding of NoLS to G-quadruplexes and interactions of NPM1wt with nucleolar proteins and rRNA.10 Ultimately, the import and anchoring signals exceed the export signals3,17 so that, at steady-state, NPM1wt resides in the nucleolus, whereas only a minimal fraction (although functionally relevant) shuttles between cell compartments.
Posttranslational modifications influence NPM1wt localization and functions
Nucleolar localization of NPM1wt depends upon its oligomerization status, because pentamers usually reside in the nucleolus, whereas monomers are nucleoplasmic.22 Phosphorylation of S48, S88, T95, and S125 favors the permanence of NPM1wt in a monomeric state, promoting its nucleoplasmic localization.23 Phosphorylation of T199, T219, T234, and T237 by a cell cycle–dependent kinase during mitosis hinders the interaction between NPM1wt and rRNA.24 We hypothesize that altering this interaction results in loss of NPM1wt phase separation, facilitating its distribution through the cell during mitosis. Acetylation of K229, K230, K257, and K267 seems to favor its nucleoplasmic localization and binding to RNA polymerase II,25 likely facilitating DNA transcription. Finally, sumoylation and ubiquitination at specific sites may influence NPM1 localization and stability.19
Structural and biological consequences of NPM1 mutations in AML
Structural consequences of NPM1 mutations
The main features of NPM1 mutations are summarized in Table 1. NPM1 mutations are always heterozygous and primarily restricted to exon 12 (<1% involving other exons).7 They probably arise from replication errors primed by an illegitimate terminal deoxynucleotidyl transferase activity.26 NPM1 mutations usually consist of 4-bp insertions that cause a frameshift in the last few C-terminal amino acids of the protein, leading to loss of W288 and W290 (or W290 alone) and generation of a new C-terminal NES27 (Figures 1A and 2B). Both changes are required for cytoplasmic localization of NPM1 mutants.3,27
Genetic features of NPM1 mutations
| Heterozygous mutations detected in about one third of adult AML (50-60% of AML with normal cytogenetic). Less frequent in children (∼7%). |
| Mostly restricted to exon 12 (<1% other exons). Mutation A (duplication TCTG) is the most common mutation (75-80% of cases), followed by mutations B and D (∼5% each). |
| Probably arising from replication errors primed by illegitimate terminal deoxynucleotidyl transferase activity. |
| Found in the whole leukemic population (as proven by IHC) and stable over time (being detected at relapse*). |
| Not detected in individuals with clonal hematopoiesis. Driver mutations acting as “gatekeepers” for AML, mostly de novo. |
| Frequently co-occur with mutations of FLT3, DNMT3A, and IDH1/2 genes. Prognosis may vary according to the associated mutations. |
| Mutually exclusive with AML entities defined by recurrent genetic abnormalities in the 2017 WHO classification of hematopoietic tumors. |
| Close association with normal karyotype (∼85% of cases). About 15% of cases carry chromosome aberrations, especially +8, del(9q), +4. |
| Associated with distinct gene expression profile characterized by upregulation of HOX genes (HOXA, HOXB) and low expression of CD34 and CD133. |
| Close association with distinct microRNA and long noncoding RNA profiles. |
| Heterozygous mutations detected in about one third of adult AML (50-60% of AML with normal cytogenetic). Less frequent in children (∼7%). |
| Mostly restricted to exon 12 (<1% other exons). Mutation A (duplication TCTG) is the most common mutation (75-80% of cases), followed by mutations B and D (∼5% each). |
| Probably arising from replication errors primed by illegitimate terminal deoxynucleotidyl transferase activity. |
| Found in the whole leukemic population (as proven by IHC) and stable over time (being detected at relapse*). |
| Not detected in individuals with clonal hematopoiesis. Driver mutations acting as “gatekeepers” for AML, mostly de novo. |
| Frequently co-occur with mutations of FLT3, DNMT3A, and IDH1/2 genes. Prognosis may vary according to the associated mutations. |
| Mutually exclusive with AML entities defined by recurrent genetic abnormalities in the 2017 WHO classification of hematopoietic tumors. |
| Close association with normal karyotype (∼85% of cases). About 15% of cases carry chromosome aberrations, especially +8, del(9q), +4. |
| Associated with distinct gene expression profile characterized by upregulation of HOX genes (HOXA, HOXB) and low expression of CD34 and CD133. |
| Close association with distinct microRNA and long noncoding RNA profiles. |
With the exception of rare cases of second AML occurring in the context of clonal hematopoiesis (see text).
Loss of tryptophan(s) causes unfolding of the C-terminal triple helix,18 impairing NPM1’s ability to reside within the nucleolus20 (Figure 2B). The new C-terminal NES27 reinforces the export activity of the N-terminal NESs,28 increasing the probability of NPM1 mutants wandering in the nucleoplasm to be exported to the cytoplasm. Importantly, XPO1 recognizes the C-terminal NES more efficiently than the N-terminal NESs, likely as a result of unfolding of the C-terminal triple helix.17 Eventually, the export exceeds the import and anchoring signals, and NPM1 mutants are delocalized to the cytoplasm of AML cells (Figure 2B).
NPM1 mutants act dominantly over NPM1wt, causing its cytoplasmic delocalization through the formation of NPM1 mutant/NPM1wt heterodimers.3,27 However, the exact proportion of NPM1wt localized to the cytoplasm of NPM1-mutated cells has never been quantified, and its biological relevance remains unclear.
NPM1 mutants are “born to be exported”
All NPM1 mutations, including those rarely occurring outside of exon 12 (ie, exon 5,29 exon 9,3 and exon 113 ) generate cytoplasmic leukemic mutants (Figure 3A). This is also a feature of rare NPM1 fusion proteins, including NPM1-HAUS1,30 and those generated by novel translocations [ie, t(5;10)(q35;q23) NPM1-RPP30, t(5;18)(q34;q12) NPM1-SETBP1, and t(5;6)(q35;q23) NPM1-CCDC28A].31 These fusion proteins lose the NPM1wt C-terminus (including W288 and W290) and acquire a C-terminal NES that is either newly created or already present in the fusion partner.
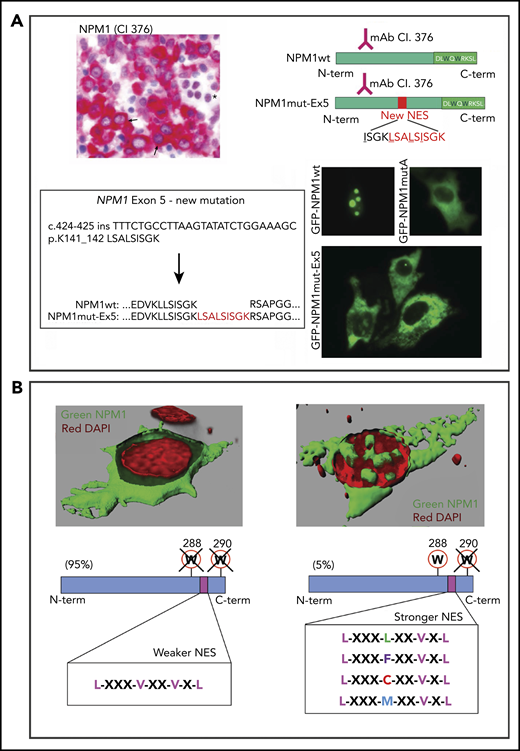
NPM1 mutants are "born to be exported." (A) A new NPM1 mutation at exon 5. IHC highlights leukemic blasts with cytoplasmic NPM1 (arrows) and normal erythroid cells with a nucleus-restricted expression of NPM1 (asterisk) (bone marrow biopsy, immune-alkaline phosphatase staining using anti–N terminus antibody, clone 376; original magnification ×400) (upper left panel). Sequence of exon 12 of NPM1 was wild-type (data not shown) (lower left panel). Targeted sequencing of the other NPM1 gene exons revealed a new mutation at exon 5, consisting of an in-frame 27-nucleotide insertion and leading to a mutant protein that is 9 aa (shown in red) longer than the wild-type (NPM1wt). Analysis of the new protein sequence predicted a newly acquired NES. Schematic representations of the new mutant protein (NPM1mut-Ex5) compared with wild-type (NPM1wt) (upper right panel). Both are recognized by the anti–N terminus monoclonal antibody (clone 376). The C terminus of the new mutant is unchanged. Cloning of the new mutant NPM1 gene sequence into a GFP-containing vector and expression in NIH3T3 cells confirmed the cytoplasmic localization of the NPM1 mutant protein (GFP-NPM1mut-Ex5), similarly to NPM1 mutant A (GFP-NPM1mutA) (lower right panel). Nucleolar localization of NPM1wt is shown as control (GFP-NPM1wt) (fluorescence microscopy, original magnification ×400). (B) Tuning of NES motif strength. When the capability of NPM1 mutant to bind the nucleolus is completely abrogated (loss of W288 and W290), a weaker C-terminal NES is inserted. When the capability of NPM1 mutant to bind the nucleolus is partially retained (loss of W290 alone), a stronger C-terminal NES motif is inserted. The 3-dimensional reconstruction in the upper left panel is from Falini et al.7
NPM1 mutants are "born to be exported." (A) A new NPM1 mutation at exon 5. IHC highlights leukemic blasts with cytoplasmic NPM1 (arrows) and normal erythroid cells with a nucleus-restricted expression of NPM1 (asterisk) (bone marrow biopsy, immune-alkaline phosphatase staining using anti–N terminus antibody, clone 376; original magnification ×400) (upper left panel). Sequence of exon 12 of NPM1 was wild-type (data not shown) (lower left panel). Targeted sequencing of the other NPM1 gene exons revealed a new mutation at exon 5, consisting of an in-frame 27-nucleotide insertion and leading to a mutant protein that is 9 aa (shown in red) longer than the wild-type (NPM1wt). Analysis of the new protein sequence predicted a newly acquired NES. Schematic representations of the new mutant protein (NPM1mut-Ex5) compared with wild-type (NPM1wt) (upper right panel). Both are recognized by the anti–N terminus monoclonal antibody (clone 376). The C terminus of the new mutant is unchanged. Cloning of the new mutant NPM1 gene sequence into a GFP-containing vector and expression in NIH3T3 cells confirmed the cytoplasmic localization of the NPM1 mutant protein (GFP-NPM1mut-Ex5), similarly to NPM1 mutant A (GFP-NPM1mutA) (lower right panel). Nucleolar localization of NPM1wt is shown as control (GFP-NPM1wt) (fluorescence microscopy, original magnification ×400). (B) Tuning of NES motif strength. When the capability of NPM1 mutant to bind the nucleolus is completely abrogated (loss of W288 and W290), a weaker C-terminal NES is inserted. When the capability of NPM1 mutant to bind the nucleolus is partially retained (loss of W290 alone), a stronger C-terminal NES motif is inserted. The 3-dimensional reconstruction in the upper left panel is from Falini et al.7
Nuclear export of NPM1 mutants is also fine-tuned by the strength of the C-terminal NES, depending on the loss of W288 and W290 (insertion of a weaker NES) or W290 only (insertion of a stronger NES)32 (Figure 3B). Rarely (eg, in exon 5 NPM1 mutations), the new NES may be strong enough to cause nuclear export of leukemic mutants without losing any of the C-terminal tryptophans (“super-NES”). Thus, all NPM1 mutations act to maximize the export of NPM1 mutants, pointing to cytoplasmic delocalization as being critical for leukemogenesis.3,32
NPM1 mutations and clonal hematopoiesis
Clonal hematopoiesis represents the expansion of a clonal population of hematopoietic precursors carrying ≥1 somatic mutation, without evidence of hematologic malignancy.33-35 Unlike mutations of genes involved in chromatin remodeling (eg, DNMT3A, TET2, ASXL1) and RNA splicing (eg, SF3B1, SRSF2), NPM1 mutations have not been detected in individuals with clonal hematopoiesis.35 Rather they act as “gatekeepers” for AML.35 A widely accepted model of clonal evolution, also supported at the single-cell level,36 sees NPM1-mutated AML developing from preexisting clonal hematopoiesis (eg, DNMT3A or IDH2 driven)33,34,37-39 (Figure 4A). Accordingly, 1 patient with IDH2-mutated clonal hematopoiesis developed AML shortly after the detection of NPM1 mutation.40 Moreover, AML patients carrying both NPM1 and DNMT3A mutations at diagnosis, who respond optimally to standard therapy, show persistence of DNMT3A mutations, despite achieving the eradication of NPM1 clone (as assessed by quantitation of NPM1-mutant transcripts) and experiencing long-term complete remission (CR) and cure.41-43 However, the return to the status of clonal hematopoiesis may predispose these patients to the development of a second hematologic neoplasm. Indeed, although most relapses in NPM1-mutated AML are due to the recurrence of the original NPM1-mutated clone, ∼5% to 10% are characterized by the absence of NPM1 mutations.44 These cases are thought to develop a second AML that is favored by the persistence of clonal hematopoiesis following the eradication of the original NPM1-mutated clone.45,46 This is supported by the long latency between CR and relapse with NPM1 wild-type AML44-46 (giving enough time for the second genetic hit to occur) and the different mutational patterns detected at diagnosis and relapse. NPM1-mutated AML may also relapse with NPM1 mutation switch (eg, from type D to A),47,48 likely representing another example of second AML arising from persistent clonal hematopoiesis. Rarely, clonal hematopoiesis may predispose to develop 2 hematological neoplasms, as we reported in a patient with angioimmunoblastic T-cell lymphoma and NPM1-mutated AML49 (Figure 4B). Thus, NPM1-mutated AML patients in CR with persistent clonal hematopoiesis should undergo long-term surveillance for the development of a second malignancy. Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the only therapy that has demonstrated the capability to eradicate clonal hematopoiesis.50
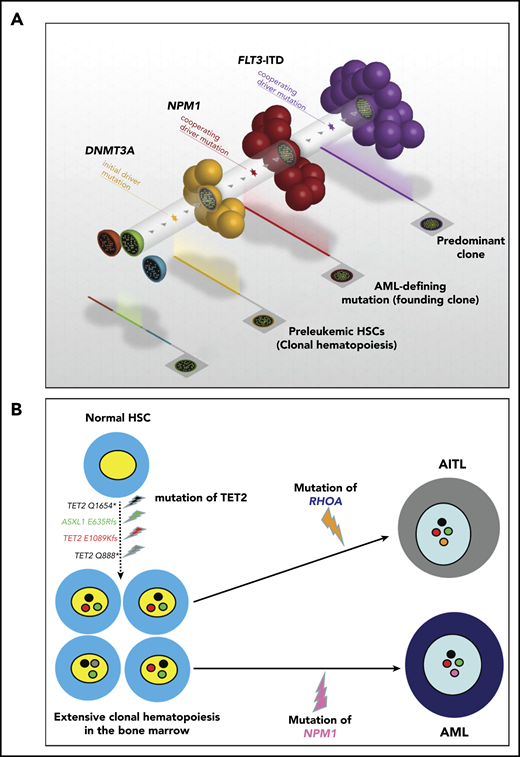
NPM1 and other mutations cooperate to promote AML. (A) AML with the triple-mutated (DNMT3A/NPM1/FLT3-ITD) genotype is observed in patients and is associated with a particularly poor outcome. DNMT3A mutations are early genetic events that are responsible for the generation of a clonal hematopoiesis. NPM1 mutations are disease-defining genetic lesions that are gatekeepers for AML, whereas FLT3-ITD mutations are late events. Courtesy of Timothy Ley. (B) Hypothetical steps in the sequential development of angioimmunoblastic T-cell lymphoma (AITL) (RHOA mutation; orange) followed by NPM1-mutated AML (NPM1 mutation; pink) in a 45-year-old man. The 2 neoplasms arose from high-risk clonal hematopoiesis in the bone marrow promoted by multiple TET2 and ASXL1 mutations.49 The TET2 Q1654* mutation (black) was present in virtually all bone marrow cells (variant allele frequency, 49.3%). The other mutations (TET2 E1089Kfs, red; TET2 Q888, gray; and ASXL1 E635Rfs, green) showing lower allele frequencies likely occurred later as subclonal events.
NPM1 and other mutations cooperate to promote AML. (A) AML with the triple-mutated (DNMT3A/NPM1/FLT3-ITD) genotype is observed in patients and is associated with a particularly poor outcome. DNMT3A mutations are early genetic events that are responsible for the generation of a clonal hematopoiesis. NPM1 mutations are disease-defining genetic lesions that are gatekeepers for AML, whereas FLT3-ITD mutations are late events. Courtesy of Timothy Ley. (B) Hypothetical steps in the sequential development of angioimmunoblastic T-cell lymphoma (AITL) (RHOA mutation; orange) followed by NPM1-mutated AML (NPM1 mutation; pink) in a 45-year-old man. The 2 neoplasms arose from high-risk clonal hematopoiesis in the bone marrow promoted by multiple TET2 and ASXL1 mutations.49 The TET2 Q1654* mutation (black) was present in virtually all bone marrow cells (variant allele frequency, 49.3%). The other mutations (TET2 E1089Kfs, red; TET2 Q888, gray; and ASXL1 E635Rfs, green) showing lower allele frequencies likely occurred later as subclonal events.
NPM1 and other gene mutations cooperate to promote AML
NPM1 and other mutations, especially those affecting FLT32 and DNMT3A,51 frequently co-occur in AML patients, implying molecular synergisms promoting AML development.52,53 Cooperation mechanisms have been investigated extensively in mouse models. Npm1/Dnmt3a double-mutated mice develop AML but only after a long latency and following transplants,38 suggesting that other mutations are necessary for AML development. Conversely, Npm1/Flt3–internal tandem duplication (ITD) double-mutated mice generate a fully penetrant and short-latency AML54,55 that is due, at least in part, to GATA1 epigenetic deregulation.56 Npm1/Flt3–tyrosine kinase domain (TKD) double-mutated mice also develop AML.57 In this model, the Npm1 mutant relocates mutated Flt3 from the cell surface to the endoplasmic reticulum, leading to Stat5 activation.57 Npm1/Flt3-ITD/Dnmt3a triple-mutated mice die of a particularly aggressive AML that is chemoresistant, likely due to impaired nucleosome remodeling.58 Accordingly, patients with this AML genotype show a very poor prognosis.53,59 These mutations synergize to drive overexpression of hepatic leukemia factor, which leads to increased self-renewal and the aberrant immunophenotype (GPR56high/CD34low) of triple-mutated cells.60,61
About 20% of NPM1-mutated AMLs carry NRAS or KRAS mutations.53 Npm1/Nras double-mutated mice develop AML with granulocytic differentiation and a less aggressive behavior than Npm1/Flt3-ITD AML, which instead shows monocytic differentiation and short latency.62,63 Similarly, patients with NPM1/NRAS double-mutated AML exhibit a relatively good prognosis.53 All of the above mouse models may serve as a platform for testing novel treatment strategies.
Does NPM1 haploinsufficiency contribute to leukemogenesis?
NPM1 mutations are always heterozygous,2 and complete loss of NPM1wt is embryonic lethal,64 implying that 1 copy of NPM1wt is necessary for cell survival. Npm1 heterozygous knock-out mice develop a myelodysplastic-like disorder that is due to uncontrolled centrosome duplication and aneuploidy,64 demonstrating that NPM1wt is an haploinsufficient tumor suppressor. However, depletion of NPM1wt in NPM1-mutated AML cells is unlikely to drive leukemia through this mechanism. Indeed, NPM1-mutated AML is closely associated with normal karyotype,7 likely because both NPM1wt and NPM1 mutants can properly regulate centrosome duplication.
Role of cytoplasmic delocalization of NPM1 mutants in AML development
NPM1 mutants induce cytoplasmic delocalization of several nuclear proteins involved in apoptosis, DNA repair and differentiation, including ARF, HEXIM1, FBW7, MIZ1, APE1, and caspases 6 and 8 (see also Falini et al3 and Heath et al9 ). Recently, altered localization of the transcription factors CTCF65 and PU.166 has been also reported (Figure 5). Overall, the degree to which the function of these proteins is disrupted by the altered localization remains to be determined because they have been mostly studied in vitro or in nonhematopoietic cells,3 and none of these NPM1 partners was confirmed to be delocalized to the cytoplasm of primary NPM1-mutated AML cells.3,67
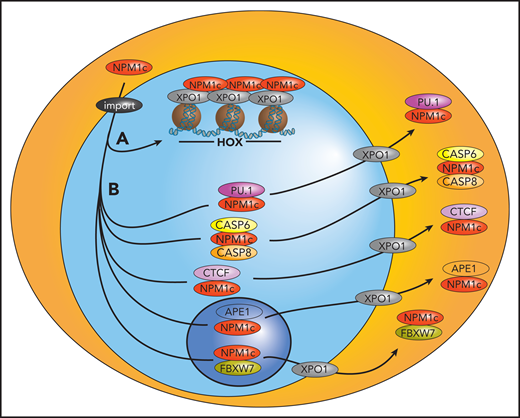
Proposed model of the pathogenesis of NPM1-mutated AML. Mutant NPM1 promotes leukemogenesis by interacting with chromatin-bound XPO1 at HOX loci to facilitate their expression (A) and binding and exporting several nuclear proteins, including tumor suppressors and transcription factors, to the cytoplasm, likely inhibiting their normal functions (B). import, importin α/β.
Proposed model of the pathogenesis of NPM1-mutated AML. Mutant NPM1 promotes leukemogenesis by interacting with chromatin-bound XPO1 at HOX loci to facilitate their expression (A) and binding and exporting several nuclear proteins, including tumor suppressors and transcription factors, to the cytoplasm, likely inhibiting their normal functions (B). import, importin α/β.
Physiologically, NPM1wt binds to a small nuclear pool of the transcriptional regulator BRD4, exerting an inhibitory effect on its transcriptional activity.68 NPM1 mutants alter this equilibrium, because a significant proportion of NPM1 is delocalized into the cytoplasm without BRD4, which is then free to drive the transcription of its target genes,68 especially BCL2 and MYC. How this mechanism contributes to AML development remains unclear.
NPM1 mutants maintain the leukemic state through HOX overexpression
HOXA and HOXB gene clusters are highly expressed in normal adult hematopoietic stem and progenitor cells (HSPCs) but are physiologically silenced in mature blood cells, suggesting a role in self-renewal.69 Moreover, ectopic HOX expression in normal HSPCs leads to leukemic transformation.69 We provided the first evidence that NPM1-mutated AML exhibits higher HOX levels (specifically HOXA and HOXB)70 as compared to NPM1wt AML. However, the HOXA and HOXB expression signature of NPM1-mutated AML is nearly identical to that of normal HSPCs, suggesting persistent expression rather than upregulation of HOX genes71 and pointing to common mechanisms regulating HOX expression in normal HSPCs and NPM1-mutated AML.71
The histone modifier MLL1 contributes by regulating HOX expression in NPM1-mutated AML, and this function depends on the interaction between MLL1 and its cofactor menin.72,73 More recently, we showed that, in NPM1-mutated AML cells, HOX expression is directly dependent on NPM1 mutants.74 Accordingly, nuclear relocalization or targeted degradation of mutated NPM1 led to immediate loss of HOX expression, followed by differentiation and growth arrest.74 Thus, NPM1 mutants act in a gain-of-function manner upstream of HOX to maintain the undifferentiated state of leukemic cells.74
The link between NPM1 mutants and HOX expression remains unclear. NPM1 could directly bind and displace putative factors from the nucleus that are required for proper differentiation and physiological HOX downregulation (eg, the myeloid transcription factor PU.166 or the architectural protein CTCF,65 although its role in HOX regulation in the context of NPM1-mutated AML has recently been questioned75 ). In this model, loss of NPM1 mutants from the cytoplasm would restore the normal activity of these factors. Alternatively, HOX expression could be maintained by recruiting NPM1 mutants to HOX loci through their interaction with chromatin-bound XPO1,76 and cytoplasmic delocalization of NPM1 would merely represent an epiphenomenon. We propose a model in which NPM1 mutants promote leukemia by acting at the chromatin level and by delocalizing NPM1-interacting partners to the cytoplasm, “killing 2 birds with 1 stone” (Figure 5).
Diagnostic and prognostic impact of NPM1 mutations
Challenges in the diagnosis of NPM1-mutated AML
NPM1-mutated AML with multilineage dysplasia77,78 (Figure 6, top panels) should be distinguished from AML with myelodysplasia-related changes (AML-MRC). According to the 2017 WHO classfication of hematopoietic tumors,5 when NPM1 mutations and multilineage dysplasia coexist, the genetic abnormality supersedes morphology, and the case is diagnosed as NPM1-mutated AML.5 Conversely, AML-MRC should be diagnosed if typical cytogenetic alterations and/or a previous history of myelodysplasia are documented, even in the presence of NPM1 mutations.5
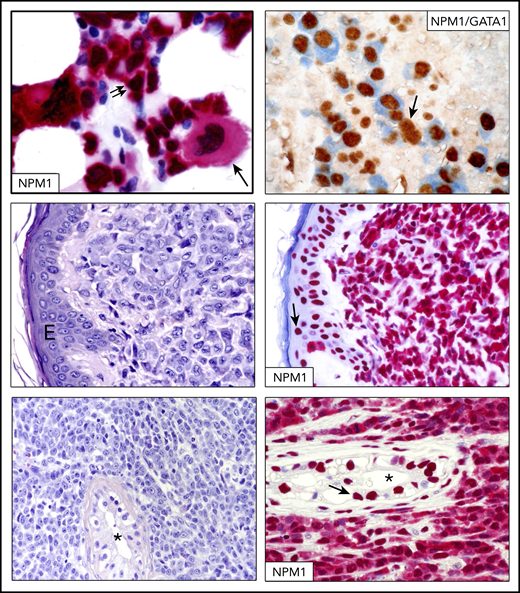
IHC in NPM1-mutated AML and myeloid sarcoma.NPM1-mutated AML with multilineage dysplasia showing infiltration by blasts (double arrows) and a monolobated megakaryocyte (single arrow) (bone marrow biopsy, immune-alkaline phosphatase staining, original magnification ×400) (top left panel). Reprinted from Pasqualucci et al.77 Another case of NPM1-mutated AML with multilineage dysplasia double stained for NPM1 (blue) and GATA1 (brown) (top right panel). Numerous erythroid leukemic cells (GATA1 nuclear brown/NPM1 cytoplasmic blue) are present. The arrow indicates a residual normal erythroid cell (original magnification ×400; double staining with immune-alkaline phosphatase/immunoperoxidase). Skin biopsy, showing dermal infiltration by leukemic cells (original magnification ×400; hematoxylin and eosin stain) (middle left panel). Leukemic cells from the same patient show aberrant cytoplasmic expression of NPM1, confirming the diagnosis of NPM1-mutated skin myeloid sarcoma (middle right panel). Normal cells of the overlying epidermis show nucleus-restricted expression of NPM1 (arrow) (original magnification ×400; immune-alkaline phosphatase staining). Testicular biopsy showing massive infiltration by leukemic cells and a residual seminiferous tubule (*) (original magnification ×400; hematoxylin and eosin stain) (bottom left panel). Leukemic cells from the same patient show aberrant cytoplasmic expression of NPM1, confirming a diagnosis of testicular myeloid sarcoma (bottom right panel). Cells of a normal residual seminiferous tubule (*) show nuclear expression of NPM1 (arrow) (original magnification ×400; immune-alkaline phosphatase staining). E, epidermis.
IHC in NPM1-mutated AML and myeloid sarcoma.NPM1-mutated AML with multilineage dysplasia showing infiltration by blasts (double arrows) and a monolobated megakaryocyte (single arrow) (bone marrow biopsy, immune-alkaline phosphatase staining, original magnification ×400) (top left panel). Reprinted from Pasqualucci et al.77 Another case of NPM1-mutated AML with multilineage dysplasia double stained for NPM1 (blue) and GATA1 (brown) (top right panel). Numerous erythroid leukemic cells (GATA1 nuclear brown/NPM1 cytoplasmic blue) are present. The arrow indicates a residual normal erythroid cell (original magnification ×400; double staining with immune-alkaline phosphatase/immunoperoxidase). Skin biopsy, showing dermal infiltration by leukemic cells (original magnification ×400; hematoxylin and eosin stain) (middle left panel). Leukemic cells from the same patient show aberrant cytoplasmic expression of NPM1, confirming the diagnosis of NPM1-mutated skin myeloid sarcoma (middle right panel). Normal cells of the overlying epidermis show nucleus-restricted expression of NPM1 (arrow) (original magnification ×400; immune-alkaline phosphatase staining). Testicular biopsy showing massive infiltration by leukemic cells and a residual seminiferous tubule (*) (original magnification ×400; hematoxylin and eosin stain) (bottom left panel). Leukemic cells from the same patient show aberrant cytoplasmic expression of NPM1, confirming a diagnosis of testicular myeloid sarcoma (bottom right panel). Cells of a normal residual seminiferous tubule (*) show nuclear expression of NPM1 (arrow) (original magnification ×400; immune-alkaline phosphatase staining). E, epidermis.
Unlike AML with recurrent genetic abnormalities, such as t(8;21), inv(16), t(16;16) or PML/RARα, ≥20% bone marrow (BM) blasts are required to diagnose NPM1-mutated AML.5 NPM1-mutated myeloid neoplasms showing ≤20% BM blasts are very rare and have been reported as myelodysplastic syndrome (MDS) or chronic myelomonocytic leukemia (CMML).79-81 However, NPM1-mutated MDS usually showed normal karyotype, CD34 negativity, and good response to chemotherapy, thus resembling AML.80,81 Similarly, NPM1-mutated CMML cases81-83 carried normal karyotype, evolved rapidly to AML,82 especially those with higher NPM1 mutation allelic burden,83 and responded better to chemotherapy than to hypomethylating agents.81 Moreover, in our experience, BM biopsies from NPM1-mutated MDS and CMML often show clusters of NPM1 cytoplasmic-positive blasts, suggestive of early AML. Whether all of the above neoplasms should be classified as NPM1-mutated AML, without regard to blast cell count,84 should be a topic for the next revision of the WHO classification.
Immunohistochemical detection of cytoplasmic NPM1 (a surrogate for NPM1 mutations85 ) may help in defining myeloid sarcoma86 (Figure 6, middle and bottom panels), which is common in NPM1-mutated AML87 and difficult to diagnose by molecular assays because of the frequent limited availability of pathological tissue. IHC also predicts NPM1 mutations outside of exon 12 that may be missed with conventional molecular techniques (Figure 3A).
Prognostic impact of NPM1 mutations
According to the 2017 European LeukemiaNet (ELN) recommendations,88 NPM1 mutations convey a relatively favorable prognosis only if FLT3-ITD is absent or shows a low allelic ratio (<0.5, FLT3-ITDlow). Three recent studies based on chemotherapy alone89-91 did not confirm the favorable prognosis of NPM1-mutated/FLT3-ITDlow AML, whereas, in the RATIFY trial, this genotype showed 73% overall survival at 5 years following chemotherapy plus the FLT3 inhibitor.92 Thus, risk stratification according to FLT3-ITD allelic ratio requires further validation depending on the treatment setting (eg, FLT3 inhibitors or allo-HSCT). The favorable impact of the NPM1-mutated/FLT3-ITD− genotype appears to be less evident in elderly patients,89 especially those older than 65 years.93 In general, adverse-risk cytogenetic abnormalities confer a poor prognosis to patients with the favorable NPM1-mutated/FLT3-ITD− genotype.94 However, these cases should be classified as AML-MRC, according to the 2017 WHO classification of hematopoietic tumors.5
NPM1-mutated AML with FLT3-ITD high allelic ratio (>0.5, FLT3-ITDhigh) and is assigned to the intermediate-risk group.88 Recently, it has been proposed that the 2017 ELN classification should be refined by moving all FLT3-ITDhigh patients, regardless of their NPM1 mutation status, from the intermediate-risk category to the adverse-risk category95 ; however, further studies are required to support this proposal. Finally, the prognostic impact of NPM1 mutant allele burden96-98 at diagnosis remains controversial.
Although the 2017 ELN classification is of practical value, genomic classification of AML has shown that the prognosis of NPM1-mutated AML may depend on a variety of accompanying mutations other than FLT3.53 For example, patients with NPM1/N-RAS, NPM1/RAD21,53 or NPM1/FLT3-TKD99 genotypes seem to have a relatively good prognosis. Conversely, cases with NPM1/WT1 double-mutated95 or NPM1/FLT3-ITD/DNMT3A triple-mutated59 genotypes show a particularly adverse outcome. A search for DNMT3A mutations has been recommended in a recent proposal to update the 2017 ELN classification.95 A limitation to the wide application of genomic classification53 is that NPM1-mutated AML is fragmented into many small molecular subsets whose prognostic impact may be difficult to study in clinical trials. Measurable residual disease (MRD) assessment in NPM1-mutated AML patients may help to overcome this limitation.
MRD assessment in NPM1-mutated AML
NPM1 mutations are ideal targets for MRD monitoring because they are frequent, stable at relapse, and not found in subjects with clonal hematopoiesis. We established a real-time quantitative polymerase chain reaction (RQ-PCR) assay to measure NPM1-mutant transcripts to predict hematological relapse and long-term survival.100 RQ-PCR remains the standard method for MRD monitoring in NPM1-mutated AML, although digital droplet PCR101,102 and NGS103,104 are emerging as alternative options. Digital droplet PCR has the advantage of a high sensitivity, and NGS can detect all types of NPM1 mutants, including those not recognized by the currently used kits for RQ-PCR. At present, both techniques are more time consuming and less cost effective than RQ-PCR, but we envision that they will become more widely used in the future.
MRD negativity measured by RQ-PCR in peripheral blood (PB)50 or BM105 and deep reduction in NPM1 mutant transcripts (>3 log in BM,106 to <1% in BM,107 or >4 log in PB108 ) at multiple time points after chemotherapy predict a low risk for leukemia relapse and good survival. Notably, MRD assessment in PB after 2 chemotherapy cycles appears to be even more accurate than genetic-based prognostication in predicting relapse risk.50
Patients in first CR who remain MRD positive after consolidation therapy benefit from allo-HSCT.108-110 However, this benefit may be only partial, because pretransplant MRD positivity predicts a poor outcome.111,112 Future studies should assess whether converting pretransplant NPM1 MRD positivity to negativity is feasible and clinically meaningful. In our experience, the risk of posttransplant leukemia relapse is reduced by using myeloablative HLA-haploidentical transplantation with regulatory and conventional T-cell–adoptive therapy.113
In general, any confirmed increase in NPM1 transcripts in BM and/or PB reliably predicts hematological relapse (usually occurring within 3 months).105,114,115 According to ELN recommendations,115 patients undergoing standard treatment should be tested by RQ-PCR at least at diagnosis, after 2 cycles of therapy, and at the end of treatment. MRD should be measured every 3 months for the first 2 years of follow-up. Then, timing of MRD monitoring should be personalized according to relapse risk. These recommendations are also applicable for the follow-up of patients undergoing allo-HSCT.
Therapy of NPM1-mutated AML: current practice and future perspectives
Current therapeutic approaches
Fit adult (≤60 years) patients with NPM1-mutated AML are treated with intensive chemotherapy. NPM1-mutated AML cells usually express high levels of CD33 that is treatable with gemtuzumab ozogamicin. Adding this agent to chemotherapy improved event-free survival and overall survival in 1 study,119 but it resulted in lower relapse incidence with no survival advantage in another study.120 In addition to chemotherapy, NPM1-mutated AML patients should receive inhibitors of FLT3, if this gene is mutated.92,121 Allo-HSCT is usually recommended for NPM1-mutated AML with FLT3-ITDhigh.88,122 Conversely, it is generally accepted that patients with NPM1-mutated AML without FLT3-ITD or with FLT3-ITDlow should not undergo allo-HSCT.92,123-125 It remains to be investigated whether the small subset of patients with low transplant–related risk, HLA-identical donor,109 and suboptimal reduction in NPM1 MRD after 2 chemotherapy cycles benefit from allo-HSCT in first CR.
Older fit patients (>60 years) with NPM1-mutated AML treated with intensive chemotherapy experience 15% to 20% long-term survival.126,127 However, these results may be ameliorated using venetoclax combined with hypomethylating agents.128 Eligible patients with NPM1-mutated AML also benefit from postremission reduced-intensity conditioning129 or nonmyeloablative130 allo-HSCT. Older unfit patients with NPM1-mutated AML rarely show long-term response to single hypomethylating agents.131 Combining venetoclax with hypomethylating agents or low-dose cytarabine is emerging as the standard of care in this setting, inducing CR in ∼90% of cases.132,133
Given the good CR rates, the National Cancer Research Institute recommends134 that, during the COVID-19 pandemic, venetoclax-based regimens should be considered as an alternative to induction chemotherapy in NPM1-mutated AML patients, particularly those aged > 50 years with comorbidities.128 Stringent molecular monitoring for NPM1 mutant transcripts is mandatory if such a strategy is adopted. This approach could help to bridge patients through the pandemic with the objective to deliver definitive therapy, including allo-HSCT, later on.
Because of the poor prognosis,81 we tend to treat patients with NPM1-mutated MDS and CMML following the same recommendations for NPM1-mutated AML; however, this issue remains controversial.
Targeting the abnormal transport of NPM1 mutants
Nuclear export of NPM1 mutants is XPO1 dependent.27 Thus, XPO1 inhibitors can redirect NPM1 mutants to the nucleus.66,74,135 However, XPO1 inhibitors are not NPM1 specific, because they also inhibit the nuclear export of other NES-containing proteins (eg, TP53 and p21).136,137 Consequently, their antileukemic activity may occur through more complex mechanisms. The selective XPO1 inhibitor selinexor shows low efficacy in AML patients,138 probably because it can be administered only once or twice a week due to its toxicity.138 This schedule does not ensure the stable XPO1 inhibition required to induce differentiation and growth arrest of NPM1-mutated AML cells139 (L.B., unpublished data, December 2019). Because of its lower blood-brain barrier penetration, the new XPO1 inhibitor eltanexor140 is better tolerated than selinexor and is currently administered up to 5 times per week in phase 1/2 studies (NCT02649790), holding promise for therapeutic success.
Targeting HOX expression
A functional HOX network is necessary to maintain the undifferentiated state of NPM1-mutated AML.62,72,74 Therefore, targeting HOX expression may prove effective in this AML entity. The histone methyltransferase MLL1 and its cofactor menin induce the trimethylation of lysine 4 on histone 3, a histone mark that correlates with active transcription of HOX genes and their cofactor MEIS1 (HOX/MEIS). Reducing HOX/MEIS expression by inhibiting menin-MLL interaction is feasible and effective in NPM1-mutated AML.72 Recently, 2 novel menin-MLL inhibitors (MI3454 and VTP-50469) exhibited striking antileukemic activity against NPM1-mutated AML in mice.73,141 Intriguingly, both inhibitors preferentially downregulated the HOX cofactor MEIS1 without impairing the expression levels of HOX genes. We believe that loss of MEIS1 may interfere with the function of multiple HOX transcription factors, leading to differentiation and growth arrest.142 Menin-MLL inhibition prevented the transformation of Npm1-mutated mouse hematopoietic progenitors into leukemic cells, implying that it could also be effective in Npm1-mutated preleukemia.73 However, we believe that menin-MLL inhibition is more likely to benefit patients with frank NPM1-mutated AML, because NPM1 mutations do not drive preleukemic clonal hematopoiesis.
Because pharmacologic XPO1 inhibition also potently downregulates HOX expression in NPM1-mutated AML,74 we envision synchronized inhibition of menin-MLL and XPO1 as an appealing therapeutic option to block HOX/MEIS expression in NPM1-mutated AML.
Targeting the nucleolus
NPM1wt is a nucleolar stress sensor,143 and the nucleolus of NPM1-mutated AML cells may be particularly vulnerable to stress because it is NPM1 depleted as a result of haploinsufficiency and cytoplasmic delocalization.7 Therefore, inducing nucleolar stress could be therapeutically effective in NPM1-mutated AML. Actinomycin D, which triggers nucleolar stress by inhibiting RNA polymerase I, induces CR in NPM1-mutated AML,144 although the mechanism of action remains unclear. Other drugs causing nucleolar stress through inhibition of ribosome biogenesis145 should be investigated in this setting. Blocking NPM1 oligomerization146 represents another potential therapeutic approach, because pentameric/decameric NPM1wt is required for proper nucleolus formation.
Mutant NPM1 as target for immunotherapy
We proposed for the first time that the unique C-terminal sequences of NPM1 mutants represent a potential immunotherapeutic target147 and that cytoplasmic localization of NPM1 mutant may favor its processing by a class I degradation pathway, leading to HLA presentation.147 This concept was further expanded demonstrating that specific autologous T-cell responses against NPM1 mutant peptides can be elicited.148-151 Immune responses were associated with molecular remission in 1 AML patient,152 and they may explain the relatively favorable outcome of NPM1-mutated AML.153 Recently, the T-cell receptor specific for 1 of these peptides bound to HLA-02*01 was isolated; following its retroviral transduction into CD4+ and CD8+ T cells, it elicited an antileukemic effect against NPM1-mutated cells.149 Thus, using engineered T cells against NPM1-mutated peptides bound to HLA represents a promising therapeutic approach.
Conclusions
NPM1 mutations are AML-driving events occurring in the setting of preleukemic clonal hematopoiesis. Evidence that eradication of NPM1-mutated clones results in AML cure41-43 (although persistent clonal hematopoiesis may require close monitoring) should encourage the development of targeted treatments for this AML entity.
Endogenous tagging of wild-type and mutated NPM174 may help us to better understand the functions of these proteins, and mass spectrometry may assist in unraveling their interactome in the cytoplasm. Clarifying how NPM1 mutants deregulate the HOX program and how HOX transcription factors control their downstream targets may also lead to new therapeutic avenues. Finally, functional genomic studies may highlight the sensitivity of combinatorial mutational events to specific drugs (eg, unexpected in vitro sensitivity to ibrutinib154 of NPM1-mutated AML cells carrying FLT3 and/or DNMT3A mutations).
Clinically, it should be clarified whether diagnosis of NPM1-mutated AML is justified, irrespective of blast percentage. Standardization of NPM1 MRD measurement, better definition of its impact at different time points, and its inclusion in the design of future clinical trials in young and elderly patients are warranted. Combining venetoclax with other drugs (eg, arsenic155 or XPO1 inhibitors)156 should be investigated further in NPM1-mutated AML.
Acknowledgments
The authors thank Ildo Nicoletti for providing the 3-dimensional reconstruction of the confocal images shown in Figure 3B and Timothy Ley (Washington University, St Louis, MO) for critically reading the manuscript and for providing Figure 4A. The authors apologize to those investigators whose papers could not be cited due to space limitations.
This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC IG 2019 number 23604 and AIRC Start-Up 2019 number 22895), the European Research Council (Advanced Grant 2016 number 740230 and Consolidator Grant 2016 number 725725), and the ARC Foundation for Cancer Research (Leopold Griffuel Prize to B.F.).
This review is dedicated to the memory of David Grimwade, who greatly contributed to establishing the clinical impact of MRD monitoring in NPM1-mutated AML.
Authorship
Contribution: B.F. coordinated the preparation of the review, and all authors performed experiments related to the review and wrote the manuscript.
Conflict-of-interest disclosure: B.F. holds a patent on NPM1 mutants (number 102004901256449). The remaining authors declare no competing financial interests.
Correspondence: Brunangelo Falini, Institute of Hematology and Centro Ricerche Emato-Oncologiche, University of Perugia, Piazzale Menghini, 9, Perugia 06131, Italy; e-mail: brunangelo.falini@unipg.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal