

Key Points
Exogenous cell-based antigen overcomes endogenous low-antigen conditions to boost virus-specific CAR T cells in vivo.
CAR T cells can control viral replication after ART withdrawal and can be reactivated by anti–PD-1 administration.

Abstract
Chimeric antigen receptor (CAR) T cells targeting CD19+ hematologic malignancies have rapidly emerged as a promising, novel therapy. In contrast, results from the few CAR T-cell studies for infectious diseases such as HIV-1 have been less convincing. These challenges are likely due to the low level of antigen present in antiretroviral therapy (ART)-suppressed patients in contrast to those with hematologic malignancies. Using our well-established nonhuman primate model of ART-suppressed HIV-1 infection, we tested strategies to overcome these limitations and challenges. We first optimized CAR T-cell production to maintain central memory subsets, consistent with current clinical paradigms. We hypothesized that additional exogenous antigen might be required in an ART-suppressed setting to aid expansion and persistence of CAR T cells. Thus, we studied 4 simian/HIV-infected, ART-suppressed rhesus macaques infused with virus-specific CD4CAR T cells, followed by supplemental infusion of cell-associated HIV-1 envelope (Env). Env boosting led to significant and unprecedented expansion of virus-specific CAR+ T cells in vivo; after ART treatment interruption, viral rebound was significantly delayed compared with controls (P = .014). In 2 animals with declining CAR T cells, rhesusized anti–programmed cell death protein 1 (PD-1) antibody was administered to reverse PD-1–dependent immune exhaustion. Immune checkpoint blockade triggered expansion of exhausted CAR T cells and concordantly lowered viral loads to undetectable levels. These results show that supplemental cell-associated antigen enables robust expansion of CAR T cells in an antigen-sparse environment. To our knowledge, this is the first study to show expansion of virus-specific CAR T cells in infected, suppressed hosts, and delay/control of viral recrudescence.
Introduction
The most successful chimeric antigen receptor (CAR) T cells described to date are directed toward antigen-abundant targets such as CD19+ leukemia cells.1-3 In contrast, anti-HIV CAR T cells are limited by insufficient viral antigen during suppressive antiretroviral therapy (ART), leading to inefficient activation, expansion, and function.4-6 CAR T cells were originally characterized as a potential therapeutic for HIV cure in human patients nearly 3 decades ago.4,5 Although these trials showed the long-term safety and persistence of infused CAR T cells, no substantive expansion or reduction in virologic status was observed.6
Recent advances in CAR T cells for the treatment of hematologic malignancies (eg, as directed against the B-cell antigen CD19) have aided in the optimization of CAR T-cell design, manufacturing, and requirements for expansion and function.2,3 Notably, CD19 CAR T-cell expansion and effector function are driven by an abundance of CD19+ tumor cells and high levels of surface-expressed antigen per cell, numbering between thousands and tens of thousands of molecules per cell depending on the leukemia.7 In stark contrast, HIV-infected cells in ART-suppressed patients are exceedingly rare, express significantly less viral antigen, and may reside predominantly in secondary lymphoid tissues, the gut, and the central nervous system.8-11 Similar barriers likely contribute to the limited success of novel CAR T-cell products directed against other malignancies, namely solid tumors.12,13
We have developed a model of ART-suppressed HIV-1 infection in rhesus macaques that is ideally suited to overcome limitations associated with low-antigen targets for CAR T-cell therapies. We combined a CD4-based CAR (CD4CAR) with CCR5 editing to protect CD4CAR T cells against simian/HIV (SHIV) infection.14-16 Our primary goal in this study was to test a combined antigen-boosting plus immune checkpoint blockade strategy designed to overcome barriers that limit CAR T cells specific for antigen-sparse targets. A secondary endpoint was to assess the efficacy of antigen-boosted virus-specific CAR T cells in infected animals following ART treatment interruption (ATI).
Methods
Ethics statement
This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health ("The Guide") and was approved by the Institutional Animal Care and Use Committees of the Fred Hutchinson Cancer Research Center/University of Washington (protocol no. 3235–06). As described previously,17 all animals were housed at and included in standard monitoring procedures prescribed by the Washington National Primate Research Center (WaNPRC), including at least twice-daily observation by animal technicians for basic husbandry parameters and daily observation by a veterinary technician and/or veterinarian. Animals were housed in cages approved by The Guide in accordance with Animal Welfare Act regulations, fed twice daily, and were fasted for up to 14 hours prior to sedation. Environmental enrichment included grouping in compound, large activity, or run-through connected cages, perches, toys, food treats, and foraging activities. If a clinical abnormality was noted, clinical veterinary staff were notified per standard WaNPRC procedures. Admission as a clinical case was solely at the discretion of clinical veterinary staff. Animals were sedated by administration of ketamine HCl and/or tiletamine/zolazepam (Telazol, Zoetis Inc., Parsippany-Troy Hills, NJ) and supportive agents before all procedures. After sedation, animals were monitored according to WaNPRC standard protocols. For minor procedures, the presence/absence of deep pain was tested by the toe-pinch reflex. The absence of response (leg flexion) to this test indicates adequate anesthesia for a given procedure. Similar parameters were used in cases of general anesthesia, including loss of palpebral reflexes (eye blink). Analgesics were provided as prescribed by clinical veterinary staff for at least 48 hours after procedures and were extended at the discretion of the clinical veterinarian based on clinical signs.
Study design and blood and tissue sampling
Animals were infected with SHIV-1157ipd3N4 via the intravenous route as previously described.18 ART was initiated 13 to 14 weeks postinfection and consisted of tenofovir disoproxil fumarate (TDF, 5.1 mg/kg), emtricitabine (FTC, 40 mg/kg), kindly provided by Gilead Sciences (Foster City, CA), and dolutegravir (DTG, 2.5 mg/kg), kindly provided by Viiv Healthcare (Research Triangle, NC).19 Following 59 to 70 weeks of suppression, animals received CD4CAR T cells, followed 19 days later by irradiated K562-Env cells.16 Twelve days after infusion of K562-Env, animals began ATI. ART was not restarted following ATI. Data presented here encompass 6 months of post-ATI follow-up; each animal continues to be monitored at the time of publication of this article. Throughout the study, peripheral blood was collected by venipuncture to monitor, for example, plasma viral loads.20 Tissues, including biopsy samples from the colon (“lower GI”), duodenum/jejunum (“upper GI”), and spleen, along with whole axillary lymph nodes and bronchoalveolar lavage, were collected at 2 and 4 weeks after infusion of CAR T cells. Lower and upper GI samples were processed as previously described.18,21 Spleen biopsy pinches were mechanically dissociated by forcing through a 70 μm filter, followed by red blood cell lysis in hemolytic buffer. Whole lymph nodes were minced and similarly filtered to obtain single-cell suspensions, which were counted and prepared for flow cytometry assays.
CAR T-cell manufacturing and infusion
Autologous T cells from each animal were collected and cryopreserved before SHIV infection (SHIV–) and after infection and ART suppression (SHIV+); SHIV– and SHIV+ cells were cultured separately throughout the CD4CAR T-cell manufacturing process. To isolate nonhuman primate (NHP) T cells, total peripheral blood mononuclear cells (PBMCs) were serially sorted by bead-based CD4-positive selection, followed by bead-based CD8-negative selection (StemCell Technologies, Vancouver, BC, Canada). Isolated CD4+ and CD8+ cells were immediately electroporated with NHP CCR5-targeted CRISPR-Cas9 ribonucleoprotein complexes, consisting of 180 pmol TrueCut Cas9 Protein v2 and 540 pmol of guide RNA (Synthego, Redwood City, CA22 ) per 2 × 107 cells. Ribonucleoproteins were incubated at room temperature for 10 minutes before mixing with cells. Cells underwent electroporation using the Lonza 4D platform, P3 Primary Cell 4D Kit L (Lonza, Basel, Switzerland), and electroporation program CY100. Cells were cultured in X-VIVO-15 media including 50 μM β-mercaptoethanol, 10% fetal bovine serum (FBS) (Gemini Bio, West Sacramento, CA), 1% penicillin/streptomycin (Thermo Fisher Scientific, Waltham, MA), 1% GlutaMAX (Thermo Fisher Scientific), and 5 ng/mL each of human interleukin-7 (IL-7) and IL-15 (PeproTech, Rocky Hill, NJ). All FBS lots were prevalidated to support robust expansion of NHP T cells in culture. NHP T cells were stimulated with an artificial antigen-presenting cell (aAPC) line engineered to express CD86 and an anti-CD3 single-chain variable fragment. aAPC media consisted of RPMI 1640 (Thermo Fisher Scientific) plus 10% FBS, 1% penicillin/streptomycin, and 1% GlutaMAX. Expanded aAPC cultures were irradiated at a dose of 100 Gy, cryopreserved, and thawed and mixed with NHP T cells at a ratio of 1 aAPC:2 T cells (using aAPC counts taken before irradiation and cryopreservation). Stimulated CD4+ and CD8+ T-cell cultures were plated separately at a concentration of 2 × 106/mL and incubated at 37°C, 5% carbon dioxide. Three days later, lentiviral vector transductions were performed by adding vectors to cells at a multiplicity of infection of 10; cells were transduced in culture media plus protamine sulfate at a concentration of 4 × 106/mL. Further information on our CD4CAR lentiviral vector can be found in the supplemental Methods (available on the Blood Web site). Following ∼4 hours of transduction at 37°C with rotation, cells were re-plated and incubated overnight at 37°C, 5% carbon dioxide. The next day, CD4+ and CD8+ cells were counted, pooled at a ratio of ∼1:1, seeded into either G-Rex10 or G-Rex100 expansion flasks (Wilson Wolf, St. Paul, MN), and then expanded for 8 days, replenishing media once on day 4. Before infusion, a small fraction of the CD4CAR T-cell product was reserved for flow cytometry– and polymerase chain reaction–based assays. For manufacturing comparison studies (Figures 1 and 2), cells were prepared identically to the infusion products, aside from the use of CD3+ selection (Stemcell Technologies), anti–CD3/CD28 magnetic beads for stimulation (Thermo Fisher Scientific), and IL-2 cytokine supplementation (PeproTech).
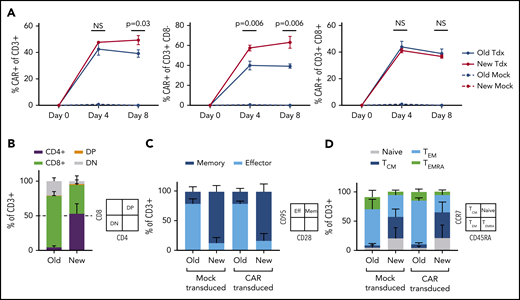
Optimized T-cell manufacturing augments CAR expression and T-cell phenotype. NHP CAR T-cell products manufactured with PBMCs from 3 uninfected animals were compared in vitro using 1 of 2 manufacturing schemes: traditional isolation of total CD3+ cells, bead-based stimulation, and culture with IL-2 (“Old”) or an updated scheme including separate isolation of CD4+ and CD8+ cells, cell-based stimulation, and no IL-2 (”New”). (A) CAR expression in total CD3+ (left), CD3+CD8– (middle), or CD3+CD8+ subsets (right). (B) CD4:CD8 ratio. (C) T-cell memory:effector ratio, determined via flow-based staining with antibodies for CD95 and CD28. (D) T-cell memory subset distribution, determined via flow-based staining with antibodies for CCR7 and CD45RA. Samples in panels B-D were collected on manufacturing day 8. Statistical significance was determined by using the Holm-Šídák method, with α = 0.05. DP, double positive; DN, double negative; NS, not significant; TCM, T central memory; Tdx, CAR-transduced; TEM, T effector memory; TEMRA, T effector memory RA.
Optimized T-cell manufacturing augments CAR expression and T-cell phenotype. NHP CAR T-cell products manufactured with PBMCs from 3 uninfected animals were compared in vitro using 1 of 2 manufacturing schemes: traditional isolation of total CD3+ cells, bead-based stimulation, and culture with IL-2 (“Old”) or an updated scheme including separate isolation of CD4+ and CD8+ cells, cell-based stimulation, and no IL-2 (”New”). (A) CAR expression in total CD3+ (left), CD3+CD8– (middle), or CD3+CD8+ subsets (right). (B) CD4:CD8 ratio. (C) T-cell memory:effector ratio, determined via flow-based staining with antibodies for CD95 and CD28. (D) T-cell memory subset distribution, determined via flow-based staining with antibodies for CCR7 and CD45RA. Samples in panels B-D were collected on manufacturing day 8. Statistical significance was determined by using the Holm-Šídák method, with α = 0.05. DP, double positive; DN, double negative; NS, not significant; TCM, T central memory; Tdx, CAR-transduced; TEM, T effector memory; TEMRA, T effector memory RA.
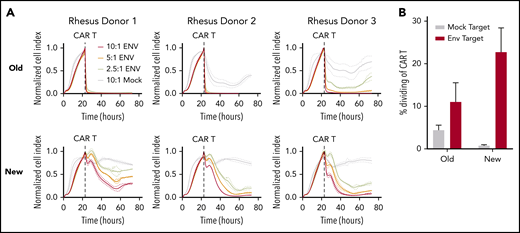
Optimized CAR T cells induce target-specific, dose-dependent cytotoxicity and proliferation. (A) Real-time cell analysis (RTCA) assay quantifying CAR T-cell cytotoxicity, using cell products from Figure 1. Target cells were the unmodified rhesus macaque kidney epithelial cell line LLC-MK2 (“Mock”) or LLC-MK2 that underwent electroporation with messenger RNA expressing the HIV-1 clade C Env from SHIV-1157ipd3N4 (“ENV”). Replicate wells containing 10:1 (red), 5:1 (orange), and 2.5:1 (yellow) ratios of CD4CAR T cells:Env targets were compared with CD4CAR T cells plated at a 10:1 ratio with Mock targets (gray) as a control. “Normalized Cell Index” is a real-time surrogate of LLCMK-2 viability, quantified via adherence to substrate (details are given in "Methods"). Target cell adherence increases until addition of CD4CAR T cells (dotted line). (B) CAR T cells were labeled with proliferation dyes during RTCA to quantify proliferation in response to mock (gray) or Env-expressing targets (red). Details of the RTCA assay are included in the supplemental Methods.
Optimized CAR T cells induce target-specific, dose-dependent cytotoxicity and proliferation. (A) Real-time cell analysis (RTCA) assay quantifying CAR T-cell cytotoxicity, using cell products from Figure 1. Target cells were the unmodified rhesus macaque kidney epithelial cell line LLC-MK2 (“Mock”) or LLC-MK2 that underwent electroporation with messenger RNA expressing the HIV-1 clade C Env from SHIV-1157ipd3N4 (“ENV”). Replicate wells containing 10:1 (red), 5:1 (orange), and 2.5:1 (yellow) ratios of CD4CAR T cells:Env targets were compared with CD4CAR T cells plated at a 10:1 ratio with Mock targets (gray) as a control. “Normalized Cell Index” is a real-time surrogate of LLCMK-2 viability, quantified via adherence to substrate (details are given in "Methods"). Target cell adherence increases until addition of CD4CAR T cells (dotted line). (B) CAR T cells were labeled with proliferation dyes during RTCA to quantify proliferation in response to mock (gray) or Env-expressing targets (red). Details of the RTCA assay are included in the supplemental Methods.
K562-Env boost
The K562-Env cell line used to boost NHP CD4CAR T cells in this study has been previously described.16 Cells were expanded in aAPC media, irradiated, and cryopreserved. Stable Env expression at the cell surface was confirmed by flow cytometry (supplemental Figure 3) using anti-HIV broadly neutralizing antibodies VRC01 and PGT126 (National Institutes of Health AIDS Reagent Program) and a polyclonal anti-immunoglobulin G phycoerythrin secondary antibody (BioLegend, San Diego, CA).23,24 For intravenous dosing in CD4CAR-treated animals, irradiated aliquots were thawed and administered at 2.5 × 107 cells per kilogram body weight, using cell counts acquired before irradiation and cryopreservation.
Rhesusized anti–programmed cell death protein 1 administration
Rhesusized anti–programmed cell death protein 1 (PD-1) (nivolumab) was acquired from the National Institutes of Health Nonhuman Primate Reagents Resource (MassBiologics, Mattapan, MA). The rhesus recombinant antibody (rhesus/human chimeric) is composed of silenced rhesus IgG4k constant regions and variable regions from the anti-human PD-1 nivolumab, and was administered intravenously at 3 mg/kg.
Statistical analyses
For comparisons of statistical significance between manufacturing schemes, an unpaired Student t test was applied between groups (n = 3), using the Holm-Šídák method, with α = 0.05. Each row was analyzed individually, without assuming a consistent standard deviation. For comparisons in time to viral rebound following ATI for untreated (n = 8) and CD4CAR T cell–treated (n = 4) animals, both two-sided Mann-Whitney (Wilcoxon rank sum test) and Grehan-Breslow-Wilcoxon tests were applied (GraphPad Prism 7, GraphPad Software, La Jolla, CA). Measures of central tendency used mean values.
Results
Optimized manufacturing conditions for low-antigen CAR T cells
In an extensive set of preliminary experiments, we prepared NHP CAR T cells that were specific for HIV-1 Env and evaluated their function in SHIV-1157ipd3N4–infected macaques. We used a manufacturing scheme previously validated in the NHP model, which closely resembles the approach used for US Food and Drug Administration–approved, cancer-specific CAR T-cell products.25 Key aspects included isolation of total CD3+ T cells, bead-based T-cell stimulation, and culture of cells in media containing IL-2. We did not observe function or expansion of these cells in vivo (C.W.P. and H.-P.K., manuscript in preparation). Rather, these cells persisted at low levels and had little or no impact on virologic parameters, similar to historical clinical studies with HIV-specific CAR T cells.4-6 To test whether virus-specific CD4CAR T cells require distinct culture conditions (eg, to maintain function in a low-antigen environment), we first compared the established manufacturing scheme with a new protocol that separately isolated CD4+ and CD8+ cells, stimulated T cells using irradiated artificial antigen-presenting cells, and omitted IL-2 from culture media.26,27 Our new manufacturing scheme supported significantly higher levels of CAR expression in vitro (Figure 1A), substantially less CD8 skewing with balanced CD4:CD8 ratios (Figure 1B), and a larger proportion of cells displaying a memory phenotype (Figure 1C), specifically central memory (Figure 1D). These cells displayed target-specific killing function that was dose dependent, whereas cells manufactured with the traditional method killed targets in a relatively binary manner (Figure 2A). Finally, expansion of cytotoxic effectors was more robust and target-specific with the new manufacturing scheme compared with the old scheme (Figure 2B). Collectively, these data suggest that our “new” manufacturing parameters support more efficient modification of T cells with CAR transgenes, increased persistence of memory subsets, and graded effector function that correlates with levels of antigen-expressing targets.
Adoptive transfer of CD4CAR T cells in nonhuman primates
Four rhesus macaques were infected with SHIV-1157ipd3N428 for 12 to 13 weeks and then placed on ART for 59 to 70 weeks (Figure 3A; supplemental Table 1) prior to infusion with CD4CAR-modified autologous T cells reprogrammed to recognize HIV-1 (Figure 3B). This SHIV strain was chosen based on our extensive previous experience17,18,29-32 and its well-established CCR5-tropism (ie, to evaluate our CCR5 editing approach).28,33 T cells from each animal were derived from 2 time points: PBMCs collected and cryopreserved before SHIV challenge (SHIV–) and freshly isolated PBMCs collected from SHIV-infected, ART-suppressed animals (SHIV+). The latter cells were included to prove that our manufacturing scheme could be applied to cells from HIV-infected, ART-suppressed patients, a key requirement for any clinically relevant CAR T-cell strategy. SHIV– and SHIV+ cells were manufactured in parallel and then mixed immediately before infusion, enabling detailed comparisons between each fraction (discussed later). Each CD4CAR T-cell product was first gene edited with CCR5 CRISPR ribonucleoprotein complexes to protect against infection with this highly CCR5-tropic SHIV15,30,34 (supplemental Figure 1). Editing of our infusion products was suboptimal (<36%). Over the first 2 months after infusion, <2% of total PBMCs were CCR5-edited and did not expand, inferring that higher levels of editing are necessary for virus-dependent positive selection. After CCR5 editing, each product underwent cell-based T-cell receptor stimulation, transduction with CD4CAR-encoding lentiviral vectors, and 8 days of expansion. CAR modification efficiency in each infusion product ranged between 20% and 50%; cells manufactured from both SHIV+ and SHIV– PBMCs were then pooled and infused intravenously into animals at a dose ranging from 2.59 × 107 to 5.92 × 107 CAR+ cells per kilogram body weight (supplemental Table 2).
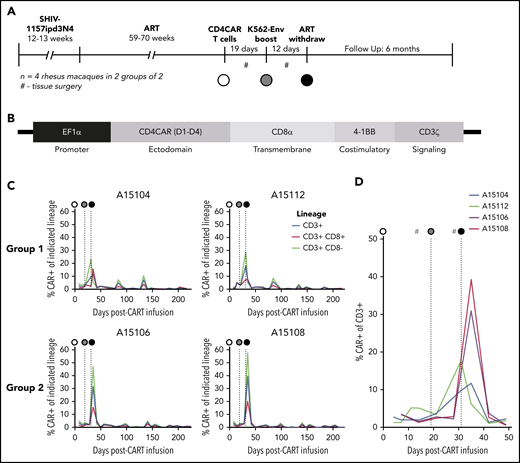
NHP CD4CAR T cells expand and persist in vivo following Env boost. (A) Experimental design for n = 4 rhesus macaques, including SHIV infection, ART suppression, CD4CAR T-cell administration (white circle), Env boost (gray circle), and ART withdrawal (black circle). (B) SIV lentiviral construct design used to deliver CD4CAR, codon optimized for rhesus macaque. (C) CD4CAR T cell frequency in peripheral blood following administration, Env boost, and ART withdrawal. Shown are total CAR T frequency of total CD3+ T cells (blue), vs CD8+ (red) and CD8– subsets (green). (D) Enlargement of total CAR T frequency of CD3+ T cell data from panel C, focused on Env boost (gray circle) and ART withdrawal (black circle). #Tissue collection, including lower and upper gastrointestinal and spleen biopsies, axillary lymph nodes, and bronchoalveolar lavage.
NHP CD4CAR T cells expand and persist in vivo following Env boost. (A) Experimental design for n = 4 rhesus macaques, including SHIV infection, ART suppression, CD4CAR T-cell administration (white circle), Env boost (gray circle), and ART withdrawal (black circle). (B) SIV lentiviral construct design used to deliver CD4CAR, codon optimized for rhesus macaque. (C) CD4CAR T cell frequency in peripheral blood following administration, Env boost, and ART withdrawal. Shown are total CAR T frequency of total CD3+ T cells (blue), vs CD8+ (red) and CD8– subsets (green). (D) Enlargement of total CAR T frequency of CD3+ T cell data from panel C, focused on Env boost (gray circle) and ART withdrawal (black circle). #Tissue collection, including lower and upper gastrointestinal and spleen biopsies, axillary lymph nodes, and bronchoalveolar lavage.
Cell-based antigen boosting of CD4CAR T cells
Preparative cytotoxic conditioning regimens are frequently administered to increase engraftment/persistence of cancer-specific CAR T cells. However, these regimens may be associated with toxicities that are unreasonable (ie, for an otherwise healthy HIV-infected individual on ART).35 To improve the safety and toxicity profile in our study, we did not administer a conditioning regimen before CD4CAR T-cell infusion. Following infusion, the absolute number and percentage of CD4CAR-modified, virus-specific effectors were quantified in the peripheral blood at serial time points. We applied the flow cytometry gating strategy shown in supplemental Figure 2, which measured CAR+ T-cell subsets on the basis of CD8 expression (using CD8– as a surrogate for CD4+), as anti-CD4 clones that label macaque CD4 but not our CD4CAR are unavailable.16 To test our primary hypothesis that an Env boost strategy could potentiate CD4CAR T cells to expand as well as recognize and kill recrudescent targets during ATI, we infused an irradiated K562 cell line modified to express HIV-1 YU2 Env (supplemental Figure 3) at day 19 following CD4CAR T-cell infusion. Twelve days following boost (31 days postinfusion of CD4CAR T cells), ART was interrupted. CD4CAR T-cell expansion was observed in all animals after Env boost and ATI, with CAR+ frequencies peaking at 20% to 50% of total peripheral T cells (Figure 3C). Notably, an immediate expansion of CD4CAR T cells in animals A15104 and A15112 was observed after Env boost, before ATI (Figure 3D); we refer to these animals as Group 1. Interestingly, the kinetics of expansion in animals A15106 and A15108 (Group 2) were delayed, but higher magnitude relative to Group 1. To our knowledge, ours is the first report of HIV/SHIV-specific CAR T-cell expansion in an autologous host.
Antigen boosting expands CD4CAR T cells in gut and secondary lymphoid tissues
To determine whether antigen boosting induced CD4CAR T-cell expansion in secondary lymphoid tissues prior to ATI and viral recrudescence, we performed tissue surgeries on each animal before and after cell-based Env boosting, prior to withdrawal of suppressive ART. Consistent with findings in the peripheral blood, total CD4CAR T cells in tissues in Group 2 did not expand initially, whereas Group 1 showed marked expansion in all tissues sampled, including gut, lymph nodes, spleen, and bronchoalveolar lavage (Figure 4A). CD4CAR T-cell expansion was observed both in the CD8+ and the CD8– fractions, with slightly increased expansion observed in the CD8– CD4CAR T cells (Figure 4B-C). Collectively, these data demonstrate expansion of HIV/SHIV-specific CAR T cells following cell-based antigen boost in peripheral blood (Figure 3) and secondary lymphoid tissues. This unprecedented finding lays the groundwork for similar applications for numerous other CAR T-cell targets with limited antigen expression.
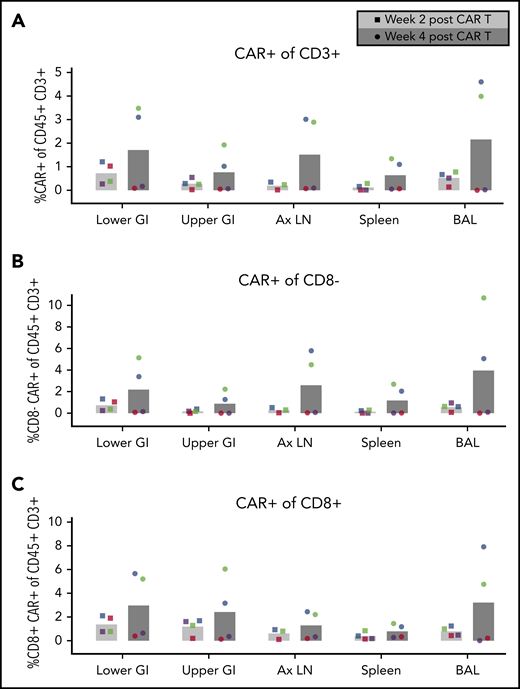
CD4CAR T-cell expansion in tissues following Env boost. (A) Total CD4CAR T-cell frequency (CD45+CD3+CAR+) in tissues before and after Env boosting. Cells from lower gastrointestinal tract (lower GI), upper gastrointestinal tract (upper GI), axillary lymph nodes (Ax LN), spleen, and bronchoalveolar lavage (BAL) were dissociated and measured for each animal (A15104, blue; A15112, green; A15106, purple; A15108, red). CD4 CAR T-cell frequency was also analyzed in CD8– (B) and CD8+ (C) T-cell subsets following Env boost at 2 weeks (light gray bars, circle symbols) and 4 weeks (dark gray bars, square symbols) after CAR T-cell infusion. Bars represent the means of n = 4 animals.
CD4CAR T-cell expansion in tissues following Env boost. (A) Total CD4CAR T-cell frequency (CD45+CD3+CAR+) in tissues before and after Env boosting. Cells from lower gastrointestinal tract (lower GI), upper gastrointestinal tract (upper GI), axillary lymph nodes (Ax LN), spleen, and bronchoalveolar lavage (BAL) were dissociated and measured for each animal (A15104, blue; A15112, green; A15106, purple; A15108, red). CD4 CAR T-cell frequency was also analyzed in CD8– (B) and CD8+ (C) T-cell subsets following Env boost at 2 weeks (light gray bars, circle symbols) and 4 weeks (dark gray bars, square symbols) after CAR T-cell infusion. Bars represent the means of n = 4 animals.
ART-free suppression of SHIV viremia after CD4CAR T-cell therapy
Although our study was primarily designed to demonstrate expansion of CAR T cells in a low-antigen setting in vivo, we also investigated the ability of expanding cells to control SHIV viremia after withdrawal of suppressive ART. Pre-ATI and post-ATI SHIV plasma viral loads were assessed in Groups 1 and 2 (Figure 5A) and compared with a cohort of 8 rhesus macaques that were infected with the same SHIV and suppressed on the same ART regimen but were otherwise untreated (supplemental Table 1). These controls displayed a mean time to viral rebound of 14.5 days (Figure 5B; supplemental Figure 4A). Following ATI, SHIV rebound in CD4CAR T-cell animals was significantly delayed relative to the control group using a rank sum test (P = .014). In particular, plasma viral load in animal A15104 remained undetectable until 89 days post-ATI. To account for potential outlier effects due to the significant delay in viral rebound in animal A15104, we also calculated percent rebound in Kaplan-Meier curves, revealing significantly delayed SHIV RNA rebound in the treatment group compared with controls using a Grehan-Breslow-Wilcoxon test (P = .02). To quantify the magnitude of viral rebound in each animal, area under the curve (AUC) of rebound SHIV plasma viremia was calculated over the same 22-week time course shown in Figure 5C. CD4CAR animals displayed lower rebound plasma viral load AUC than the control group, but due to the well-characterized variability in SHIV viral set points,30,36,37 these trends did not reach statistical significance (supplemental Figure 4B). Intriguingly, Group 1 animals exhibited 2 to 3 viral blips after ATI that temporally correlated with transient CAR T-cell expansion, whereas Group 2 animals failed to control the virus long term, despite clear evidence of post-ATI CD4CAR T-cell expansion. At the time of manuscript submission, plasma viral loads in animal A15112 have stabilized in the range of 102 copies/mL, whereas A15104 continues to display occasional low-level viral blips and recontrol. Collectively, these results indicate that CD4CAR T-cell therapy coupled with cell-associated Env boost supports ART-free suppression of SHIV viremia to 102 copies or less in 2 of 4 treated animals; these animals (Group 1) also showed the most immediate response to antigen boosting.
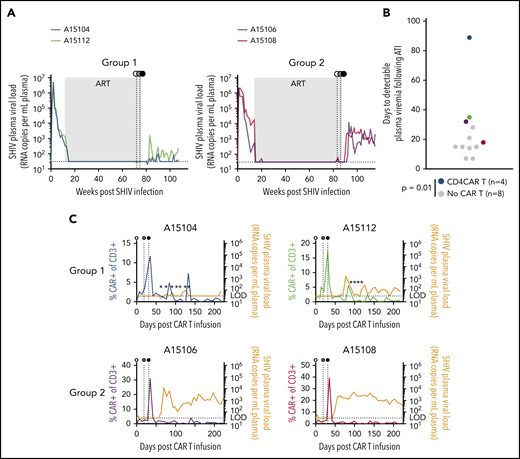
ART-free suppression of SHIV viremia following CD4CAR T-cell therapy. In each panel, animal A15104 is blue, A15112 is green, A15106 is purple, and A15108 is red. (A) Longitudinal plasma viral loads in animal A15104 and animal A15112 (Group 1) and animal A15106 and animal A15108 (Group 2). Gray boxes represent the >1 year duration of ART suppression. (B) Time to detectable SHIV rebound after ART withdrawal. Colored circles correspond to color codes in panel A. Black circles represent control animals that were infected with the same SHIV and suppressed for >1 year but did not receive CAR T cells or Env boost (P = .014 calculated by using the Mann-Whitney test). (C) Overlays of plasma viral load and total CAR T-cell frequency for each animal. Stars indicate plasma viral load values detected only in 1 of 2 assay replicates, and horizontal dotted line indicates limit of detection for plasma viral load assay (30 copies/mL). Detectable rebound is defined as a sample with a value >30 copies/mL in both replicate polymerase chain reaction reactions. In panels A and C, white circles represent CD4CAR T-cell administration; gray circles, Env boosting; and black circles, ART withdrawal.
ART-free suppression of SHIV viremia following CD4CAR T-cell therapy. In each panel, animal A15104 is blue, A15112 is green, A15106 is purple, and A15108 is red. (A) Longitudinal plasma viral loads in animal A15104 and animal A15112 (Group 1) and animal A15106 and animal A15108 (Group 2). Gray boxes represent the >1 year duration of ART suppression. (B) Time to detectable SHIV rebound after ART withdrawal. Colored circles correspond to color codes in panel A. Black circles represent control animals that were infected with the same SHIV and suppressed for >1 year but did not receive CAR T cells or Env boost (P = .014 calculated by using the Mann-Whitney test). (C) Overlays of plasma viral load and total CAR T-cell frequency for each animal. Stars indicate plasma viral load values detected only in 1 of 2 assay replicates, and horizontal dotted line indicates limit of detection for plasma viral load assay (30 copies/mL). Detectable rebound is defined as a sample with a value >30 copies/mL in both replicate polymerase chain reaction reactions. In panels A and C, white circles represent CD4CAR T-cell administration; gray circles, Env boosting; and black circles, ART withdrawal.
Ex vivo correlates of in vivo CD4CAR T-cell function
To gain insight into potential differences between Groups 1 and 2, we interrogated the phenotype of each CD4CAR T-cell infusion product using markers of cellular proliferation, activation state, memory subset, and coinhibitory molecule expression. Group 1 animals had slightly increased CAR expression, particularly in the CD8– lineage; CAR modification of SHIV+ cells was only modestly decreased relative to SHIV– cells collected prior to infection (Figure 6A). Notably, we detected only low levels of SHIV DNA and SHIV RNA in the SHIV+ fraction of each infusion product, and these levels did not significantly increase during the manufacturing process (supplemental Figure 5). During manufacturing, Group 1 CD4CAR T-cell products showed decreased cellular proliferation (Ki67), increased activation (CD69), and decreased expression of coinhibitory molecules (PD-1, TIGIT [T-cell immunoreceptor with immunoglobulin and ITIM domains]), relative to animals in Group 2 (Figure 6B-D). There were no obvious differences between the groups in terms of CD28 or CD95 expression, with each infusion product strongly skewed toward a memory phenotype. Expression of the memory subset markers CCR7 and CD45RA was also comparable between the groups, with a majority showing an effector memory phenotype (Figure 6E-F). In sum, lower levels of CD4CAR T-cell proliferation and higher levels of activation ex vivo could potentially account for the control of virus in Group 1, but not Group 2, consistent with our in vivo data.
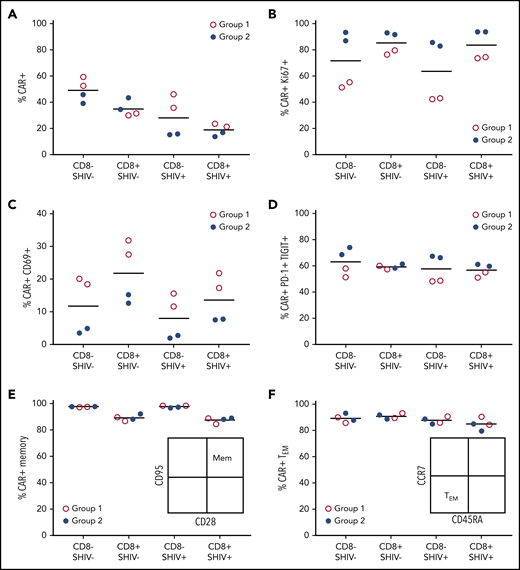
CD4CAR T-cell infusion products reveal correlates of successful Env boosting. Phenotypic differences in infusion products for groups 1 (A15104, A15112, red) and 2 (A15106, A15108, blue) were assessed by flow cytometry 8 days following after transduction, immediately before in vivo administration. (A) CAR+ cells. (B) CAR+Ki67+ cells. (C) CAR+CD69+ cells. (D) CAR+PD-1+TIGIT+ (T-cell immunoreceptor with Ig and ITIM domains) cells. (E) CD95+CD28+ memory CAR T cells. (F) CCR7–CD45RA+ effector memory CAR T cells. CD8–, surrogate for CD4+ T cells; SHIV–, cells collected and cryopreserved before SHIV infection; SHIV+, cells collected with or without cryopreservation, following SHIV infection and stable suppression by ART.
CD4CAR T-cell infusion products reveal correlates of successful Env boosting. Phenotypic differences in infusion products for groups 1 (A15104, A15112, red) and 2 (A15106, A15108, blue) were assessed by flow cytometry 8 days following after transduction, immediately before in vivo administration. (A) CAR+ cells. (B) CAR+Ki67+ cells. (C) CAR+CD69+ cells. (D) CAR+PD-1+TIGIT+ (T-cell immunoreceptor with Ig and ITIM domains) cells. (E) CD95+CD28+ memory CAR T cells. (F) CCR7–CD45RA+ effector memory CAR T cells. CD8–, surrogate for CD4+ T cells; SHIV–, cells collected and cryopreserved before SHIV infection; SHIV+, cells collected with or without cryopreservation, following SHIV infection and stable suppression by ART.
Immune exhaustion contributes to suboptimal CD4CAR T-cell function
We next looked for SHIV mutations in the Env open reading frame in circulating virus from Group 2 animals, which could have enabled viral escape from CD4CAR T cells. No relevant mutations (eg, in the CD4 binding site) were observed in either animal (supplemental Figure 6). We hypothesized that the lack of viral control was instead due to CAR T-cell exhaustion, consistent with the increased expression of the coinhibitory molecules PD-1 and TIGIT on the Group 2 infusion products (Figure 6D). To directly test whether CAR T cells were exhausted in vivo, we treated the Group 2 animals with a rhesusized version of the anti–PD-1 checkpoint inhibitor nivolumab at 227 days after CAR T-cell infusion. We opted not to treat Group 1 animals, because both were still sporadically controlling viremia to low or undetectable levels at this time. We first confirmed that this antibody bound the PD-1 receptor in peripheral T-cell populations by comparing PD-1 occupancy before and after nivolumab administration, using a mean fluorescence intensity–based approach. Consistent with our prediction, CD8+ CAR T cells displayed the highest levels of PD-1 expression in vivo before treatment and were most dramatically affected following nivolumab dosing and receptor blockade (Figure 7A). The impact of PD-1 blockade on total peripheral T cells was modest (Figure 7B). In stark contrast, anti–PD-1 selectively increased the frequency of CD4CAR T cells, with CD8+ CAR T cells expanding specifically (Figure 7C). Transient viral control was observed in both Group 2 animals after checkpoint inhibitor treatment (Figure 7D). Virus rebounded shortly after nivolumab washout. Despite a subsequent increase in circulating viral antigen, peripheral CAR T cells did not expand (Figure 7E). Collectively, these data show that lack of function of CAR T cells in the Group 2 animals was associated with a persistent immune exhaustion phenotype, which was transiently released by the PD-1 immune checkpoint blockade.
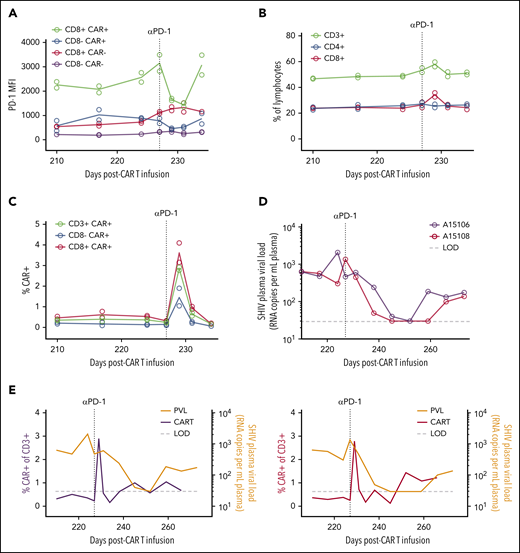
Reversal of CAR T-cell exhaustion following anti–PD-1 therapy. (A) PD-1 occupancy in CAR–positive and –negative subsets after anti–PD-1 administration on day 227 post–CAR T-cell infusion (vertical dotted line). (B) Frequency of total T-cell subsets after anti–PD-1. (C) Frequency of CAR T-cell populations after anti–PD-1. In panels A and C, circles represent individual animals (A15106 and A15108), and lines represent means (n = 2). (D) Longitudinal plasma viral loads after anti–PD-1. (E) Overlays of plasma viral load and total CAR T-cell frequency from panels C-D. LOD (horizontal dotted line), limit of detection for plasma viral load assay (30 copies/mL).
Reversal of CAR T-cell exhaustion following anti–PD-1 therapy. (A) PD-1 occupancy in CAR–positive and –negative subsets after anti–PD-1 administration on day 227 post–CAR T-cell infusion (vertical dotted line). (B) Frequency of total T-cell subsets after anti–PD-1. (C) Frequency of CAR T-cell populations after anti–PD-1. In panels A and C, circles represent individual animals (A15106 and A15108), and lines represent means (n = 2). (D) Longitudinal plasma viral loads after anti–PD-1. (E) Overlays of plasma viral load and total CAR T-cell frequency from panels C-D. LOD (horizontal dotted line), limit of detection for plasma viral load assay (30 copies/mL).
Discussion
CAR T-cell therapies for HIV have so far lagged behind those for cancer.6 Consistent with waning virus-specific T-cell responses during ART-dependent viral suppression,38 it is reasonable to assume that a lack of requisite antigen contributes to inefficient virus-specific CAR T-cell recognition/killing of infected cells and trafficking to tissue sites.39,40 Similar shortcomings with lack of persistence and expansion have been observed by groups infusing expanded modified or natural cytotoxic T lymphocytes specific to HIV, particularly related to immune-mediated clearance or apotosis.41-43
Our preliminary studies in the NHP model indicated that nonboosted virus-specific CAR T cells did not expand or affect SHIV viremia in vivo but did persist at low levels, consistent with early clinical findings (manuscript in preparation). 4-6 Here, we have implemented a novel strategy to introduce exogenous antigen to aid in CAR T-cell expansion and persistence. We optimized our CAR T-cell manufacturing to maintain a central memory phenotype with balanced CD4:CD8 T cell ratios to aid engraftment and persistence in vivo. After infusion of optimized CD4CAR T cells, we show for the first time robust expansion of anti-HIV CAR T cells in the macaque model following in vivo modulation by a cell-based Env boost. CAR T cells expanded in all 4 animals following boosting. Interestingly, expansion in one pair of animals (referred to as Group 1) was earlier and lower in magnitude than in the other pair (Group 2), in which the magnitude of expansion was later and substantially higher. Intriguingly, the Group 1 animals that displayed “slow burn” kinetics of CD4CAR expansion were able to control viral replication with Env boosting alone, whereas viral control was not initially observed in Group 2 animals that exhibited a “short burst” of CD4CAR expansion. We postulate that a slow burn model (ie, potent antiviral activity over a prolonged time period even at lower levels) is important to target and clear latently infected cells, which may recrudesce over an extended time frame.44-46
A common challenge in CAR cell therapy for malignancies is T-cell exhaustion, which can be overcome both intrinsically47,48 and extrinsically with immune checkpoint blockade.49 ART-suppressed persons living with HIV with associated hematologic malignancies have been safely treated with checkpoint inhibitors,50-54 although little is known about the impact of these therapies on viral persistence. Numerous studies in the NHP model collectively suggest that checkpoint blockade does indeed augment the endogenous T-cell response and provides clinical benefit both in prophylactic and therapeutic models.55-60 Based on these findings, we administered an anti–PD-1 checkpoint inhibitor to test whether T-cell exhaustion was a factor in the 2 animals that did not control virus long term. After treatment, we observed CAR T-cell expansion and transient viral control to undetectable levels. These data suggest that exhausted anti-HIV CAR T cells can be “rescued” with checkpoint inhibitors, a combinatorial approach that is already under investigation for cancer-specific CAR T cells in clinical trials.61
Our findings are consistent with the hypothesis that antigen supplementation may overcome challenges associated with the recognition of low-antigen targets. In the case of HIV/SHIV infection, antigen boosting may effectively prime virus-specific CAR T cells before ATI, allowing these cells to stay ahead of recrudescent viremia, while also allowing them to escape exhaustion in response to recrudescent virus. The ongoing control of virus in 2 animals lends credence to our approach as being a reasonable strategy to achieve durable ART-free remission of HIV-1 in infected patients, whereas the other 2 animals highlight the ability of exhausted anti-HIV CAR T cells to respond favorably to checkpoint blockade following exhaustion. This is the first report of exogenous antigen boosting and immune checkpoint blockade to expand anti-HIV CAR T cells in an NHP model. The clinical implementation of our K562 cell line–based antigen boosting approach could present regulatory challenges. However, analogous approaches to deliver Env antigens (eg, utilizing various nanoparticle-based strategies) have shown marked successes in recent clinical trials (reviewed by Anselmo and Mitragotri62 ). Furthermore, using recombinant protein and RNA-based antigen delivery, similar boosting approaches have recently been described for CAR T cells directed against solid tumors in mouse models.25,63 Our results are consistent with these data, indicating that similar strategies may also be required for malignant targets in which insufficient antigen is a primary barrier to efficacy.64-66 Additional research will also be necessary to merge immune checkpoint blockade with CAR T-cell therapies in clinical studies. Clinical trials combining cancer-specific CAR T cells and immune checkpoint blockade require substantially greater study. The reversal of T-cell exhaustion following antibody-based PD-1 blockade in our study was transient and may not protect against latently infected cells that recrudesce months or years after withdrawal of suppressive ART. Notably, further development of CAR T-cell gene-editing approaches (eg, to directly inactivate PD-1 expression) are highly promising and feasible in patients.67
We and others have previously established proof of principle that the curative approaches applied to formerly HIV-infected patients in Berlin and London included a component of enhanced virus-specific immunity.29,68,69 Importantly, both patients experienced toxicities that are only applicable for those with HIV-associated malignancies. Likewise, CAR T-cell therapies for cancer frequently require aggressive and often toxic conditioning regimens to maximize CAR T-cell engraftment, followed by risks associated with cytokine release syndrome during CAR T-cell expansion in vivo.70 A primary goal for virus-specific CAR T-cell therapies in otherwise healthy persons living with HIV-1 infection is to provide enhanced virus-specific immunity with minimal toxicity. Our study accomplished this goal in 3 respects. First, CD4CAR T cell–treated animals did not receive a preparative conditioning regimen before infusion of CD4CAR T cells. Second, CD4CAR T-cell infusion was well tolerated, with no evidence of cytokine release syndrome or neurotoxicities that have been observed previously in the monkey model.25 Finally, we saw no nonspecific binding of the CD4CAR to endogenous MHCII molecules, a safety concern that has also been alleviated in clinical CD4CAR T-cell trials.71,72 In short, the safety profile of our virus-specific CAR T-cell therapy is extremely favorable, supporting an ongoing clinical trial of CD4CAR T cells in HIV-infected, stably suppressed patients (ClinicalTrials.gov #NCT03617198).
In summary, we have developed a robust large animal model of CAR T-cell therapy using clinically relevant manufacturing schemes and methods, supporting expansion and function of CAR-modified effectors in the peripheral blood as well as in tissues. We show that virus-specific CAR T cells can be expanded both by rationally designed cell-based antigens and by immune checkpoint blockade. This study shows the feasibility of our approach not only for HIV but potentially also for liquid and solid tumors in which low antigen expression is a key limitation.
All data supporting the findings of this study are available within the paper and its supplemental information files, or from the corresponding authors upon request.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Helen Crawford for assistance in preparing the manuscript; Veronica Nelson, Erica Curry, Kelvin Sze, Megan Brown, Sarah Herrin, and Michelle Hoffman for outstanding support in the NHP studies; and Gilead Sciences and Viiv Healthcare for providing ART drugs. The authors are grateful to Aimee Payne for kindly providing the K562-Env cell line. They also thank Patricia Firpo and Sandra Dross for plasma viral load assays; Solomon Wangari, Britni Curtis, and WaNPRC staff for tissue collections; Teresa Einhaus, Dnyanada Pande, and Rasika Venkataraman for CCR5 editing assays; and Semih Tareen and James Hoxie for helpful discussions. The following reagents were obtained through the AIDS Reagent Program, Division of AIDS, National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID): anti–HIV-1 gp120 Monoclonal (VRC01), from Dr. John Mascola (catalog #12033), and anti–HIV-1 gp120 Monoclonal (PGT126) from IAVI (catalog #12344). Anti–PD-1 reagents used in these studies were provided by the NIH Nonhuman Primate Reagent Resource (R24 OD010976, U24 AI126683).
This study was supported by grants from the NIH/National Heart, Lung, and Blood Institute (U19 HL129902, H.-P.K. and L.S.K.) and NIH/NIAID (UM1 AI126623, K.R.J. and H.-P.K.; UM1 AI126617, R33 AI116184, and U19 AI117945, L.S.K.; UM1 AI126620 and U19 AI117950, J.L.R.; and U19 AI149680 to H.-P.K and J.L.R.). UM1 AI126620 (J.L.R.) is cofunded by NIH/NIAID, NIH/National Institute of Neurological Disorders and Stroke, and NIH/National Institute on Drug Abuse. C.R.M. is supported by a T32 grant (AI00763). The AIDS and Cancer Virus Program (C.M.F. and B.F.K.) is funded, in part, with federal funds from the NIH/National Cancer Institute (75N91019D00024).
Authorship
Contribution: H.-P.K. and C.W.P. are the principal investigators of the study and designed and coordinated the overall execution of the project; C.W.P. and B.J.R. designed the animal experiments; L.S.K. and L.C. established suppressed SHIV infection in each animal and provided feedback on NHP CAR T-cell experiments; K.E.B. cultured and prepared irradiated K562-Env cells with feedback from C.R.M., G.I.E., and J.L.R.; L.C., K.E.B., N.H.P., and W.O. collected and processed longitudinal samples and performed flow cytometry assays, which were analyzed by B.J.R.; K.E.B. and N.H.P. manufactured the CAR T-cell products, using a protocol developed by B.J.R. and C.W.P., with feedback from C.R.M., G.I.E., and J.L.R.; C.M.F. and B.F.K. generated and analyzed viral sequencing data; M.R.E., B.J.R., and C.W.P. performed CCR5 editing and SHIV plasma viral load AUC analyses; M.-L.H. and K.R.J. quantified cell-associated SHIV in CAR T-cell infusion products and sorted PBMCs; and B.J.R., H.-P.K., and C.W.P. wrote the manuscript.
Conflict-of-interest disclosure: J.L.R. cofounded a company called Tmunity Therapeutics that has the rights to license the technology described in this paper; and he holds an equity interest in Tmunity. The remaining authors declare no competing financial interests.
Correspondence: Hans-Peter Kiem, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Mail Stop D1-100, Seattle, WA 98109-1024; e-mail: hkiem@fhcrc.org; or Christopher W. Peterson, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave N, Mail Stop D1-100, Seattle, WA 98109-1024; e-mail: cwpeters@fhcrc.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal