

Key Points
miR-185 is discovered to predict therapy response in treatment-naive patients with CML, and functions as a tumor suppressor.
Its restored expression impairs LSC survival, sensitizing them to therapy; PAK6 is a target gene of miR-185, mediating drug resistance.

Abstract
Overcoming drug resistance and targeting cancer stem cells remain challenges for curative cancer treatment. To investigate the role of microRNAs (miRNAs) in regulating drug resistance and leukemic stem cell (LSC) fate, we performed global transcriptome profiling in treatment-naive chronic myeloid leukemia (CML) stem/progenitor cells and identified that miR-185 levels anticipate their response to ABL tyrosine kinase inhibitors (TKIs). miR-185 functions as a tumor suppressor: its restored expression impaired survival of drug-resistant cells, sensitized them to TKIs in vitro, and markedly eliminated long-term repopulating LSCs and infiltrating blast cells, conferring a survival advantage in preclinical xenotransplantation models. Integrative analysis with mRNA profiles uncovered PAK6 as a crucial target of miR-185, and pharmacological inhibition of PAK6 perturbed the RAS/MAPK pathway and mitochondrial activity, sensitizing therapy-resistant cells to TKIs. Thus, miR-185 presents as a potential predictive biomarker, and dual targeting of miR-185-mediated PAK6 activity and BCR-ABL1 may provide a valuable strategy for overcoming drug resistance in patients.
Introduction
One of the greatest barriers to treating cancer is drug resistance.1 In leukemia, this is primarily because of the inability of available therapeutics to eradicate a unique subset of persisting drug-resistant cells, with stem cell properties and the unique ability to regenerate disease recurrence.2,3 Imatinib mesylate (IM) and other BCR-ABL1 tyrosine kinase inhibitors (TKIs) are among the first examples of highly effective therapeutics that specifically target the kinase activity encoded in the BCR-ABL1 fusion gene in patients with early-phase chronic myeloid leukemia (CML).4-7 However, TKI monotherapies are generally not curative, as most patients harbor residual leukemic stem cells (LSCs), and disease usually recurs if TKI therapy is discontinued.8,9 In fact, LSCs (and their progenitors) are relatively insensitive to TKIs and are genetically unstable, enabling aggressive subclones to emerge over time.3,10-12 Treatment of resistant chronic or accelerated phase CML, blast crisis CML, and BCR-ABL1+ acute lymphoblastic leukemia (ALL), which closely resembles the lymphoid blast crisis of CML, pose even greater challenges, as TKI monotherapy is less effective.13-16 Allogeneic transplants remain the only curative therapy, but the associated risk for mortality and morbidity, restrictions to younger patients, and a lack of suitable donors limit their utility.17 Therefore, predictive biomarkers and novel therapeutic approaches are clearly needed.
The discovery of microRNAs (miRNAs) and their role in regulating normal physiological processes and in the pathogenesis of human cancers has been a revolutionary development.18 miRNAs are small, noncoding, single-stranded RNAs of 18 to 25 nucleotides that control gene expression by destabilizing target transcripts and inhibiting their translation.18 They play a key role in regulating multiple biological processes, including cell proliferation, survival, and differentiation in many tissues, including the process of hematopoietic cell production.19-21 Aberrantly expressed miRNAs that act as tumor promoters or suppressors have been implicated in many diseases, including cancer.22,23 The ability of miRNAs to target multiple genes and signaling pathways has also created immense interest in their utility as predictive and diagnostic biomarkers, and as innovative therapeutic agents.24,25 In human acute myeloid leukemia, miRNAs have already been identified and found to correlate with risk categories and progression.24,26-28 In CML, miRNA expression profiling or target gene predictions have been used to identify miRNAs that directly target BCR-ABL129,30 and miRNAs that are regulated by BCR-ABL1.31,32 However, it has been difficult to reach a consensus on miRNA signatures because of the diversity of samples and analysis platforms used, with most studies based on extracts of cell lines or unselected cells from patients at different stages of disease or of unknown TKI response status. Therefore, the question of how deregulated miRNAs and their target genes might contribute to the pathogenesis or drug response of patients’ primitive CML cells remains poorly understood.
To address this issue, we generated global transcriptome profiles on purified subsets of cells from patients with CML with known subsequent TKI responses. Analysis of these data sets identified miR-185 as one of the most deregulated miRNAs, with significant reduction in CD34+ cells from TKI nonresponders compared with TKI responders. PAK6 was uncovered as a target gene of miR-185, with inversely correlated expression, mediating drug resistance in TKI nonresponder cells. Further studies provided new insights into how this information might predict patient responses to therapy and improve the treatment of CML and BCR-ABL1+ ALL.
Methods
Human cells
Heparin-anticoagulated peripheral blood (PB) or bone marrow (BM) cells were obtained from 22 (cohort 1) or 58 (cohort 2) newly diagnosed patients with CML-chronic phase at diagnosis, before initiation of IM or nilotinib (NL) therapies (supplemental Table 1, available on the Blood Web site; CAMN107E2401-ENESTxtnd phase IIIb clinical trial, ClinicalTrials.gov identifier: NCT01254188).33 Patients were later classified as TKI responders or nonresponders according to the European Leukemia Net treatment guidelines.34-36 Additional samples were obtained 1 and 3 months posttreatment in the second cohort (116 samples). Normal BM (NBM) samples from healthy adult donors were obtained from the Hematology Cell Bank of British Columbia. Informed consent was obtained in accordance with the Declaration of Helsinki, and the procedures used were approved by the Research Ethics Board at the University of British Columbia. CD34+ cells (>90%) were enriched immunomagnetically, using EasySep CD34 selection kits (STEMCELL Technologies) and analyzed using a fluorescence-activated cell sorter (FACS). The BCR-ABL1+ human cell lines studied included K562, an IM-resistant K562 derivative (K562R), BV173 cells, BCR-ABL1-transduced UT7 cells (UT-B/A), and BCR-ABL1-T315I-transduced cells (UT-B/A-T315I).37,38
microRNA and strand-specific RNA sequencing and bioinformatics analyses
Total RNA (1 µg per sample) was extracted, fractionated, ligated, and converted to cDNA (Invitrogen). RNA libraries were made and sequenced. The Bioconductor DESeq2 package was used to rank differentially expressed miRNAs.39 Differentially expressed miRNAs were clustered and visualized in heat maps, using hierarchical clustering and the heat map function in R (default parameters). Strand-specific RNA sequencing and bioinformatics analysis are detailed in the supplemental Methods.
Microfluidic quantitative polymerase chain reaction and bioinformatics analysis
This method has been previously described.40 Briefly, total RNAs were extracted and 25 ng RNA of each sample was then reverse transcribed into cDNA. Preamplification, microfluidic quantitative polymerase chain reaction (qPCR), and bioinformatics analysis were performed. Additional details are provided in supplemental Methods.
Inhibitors
IM, dasatinib (DA), NL, and PF-3758309 were obtained from Novartis, Bristol-Myers Squibb, or Selleckchem. Stock solutions of 10 mM were prepared with water (IM) or dimethyl sulfoxide (DA, NL, and PF-3758309) and stored at −20°C.
Colony-forming cell and long-term culture-initiating cell assays
Colony-forming cell (CFC) and long-term culture-initiating cell (LTC-IC) assays were performed as previously described.41 For replating assays, cells from primary CFC assays were harvested, washed once with phosphate-buffered saline, and replated in fresh methylcellulose assay medium of the same composition used for the primary assay, and colonies were counted.
Xenotransplantation experiments
BV173YFP/Luc cells (2.5 × 106 per mouse) were injected into nonobese diabetic/severe combined immunodeficiency interleukin-2 receptor gamma chain (IL2Rγ)–deficient mice, treated with DA, and analyzed as described.42 CD34+ CML cells (2 × 106 cells per mouse), transduced with a control vector or the miR-185 expressing vector, were cultured with or without DA (75 nM) for 24 hours or injected intravenously into nonobese diabetic-Rag1−/−IL2Rγc−/− mice. PB and BM samples were collected, and mice were euthanized and analyzed 25 weeks posttransplantation (additional details in supplemental Methods). Animal experiments were performed in the Animal Resource Centre of the BC Cancer Research Institute, using procedures approved by the Animal Care Committee of the University of British Columbia (Vancouver).
Data analysis
Results are shown as the mean and standard error of the mean from at least 3 independent experiments. Differences between groups were evaluated using the 2-tailed Student t test or 1-way analysis of variance with post hoc testing for multiple comparisons. Additional details are available in supplemental Methods.
Results
Deep sequencing identifies miR-185 as a biomarker of TKI response in treatment-naive CML stem/progenitor cells
To identify differentially expressed miRNAs that might serve as biomarkers and/or therapeutic targets, we performed Illumina deep sequencing on CD34+ cells from 6 patients with CML at diagnosis (supplemental Figure 1). Three of the patients were subsequently classified clinically as IM responders, and 3 as IM nonresponders (supplemental Table 1).35 CD34+ cells isolated from NBM samples from 3 healthy donors were used as controls. Bioconductor DESeq2 analysis revealed 66 differentially expressed miRNAs between CML and NBM samples (with adjusted P value < .05; Figure 1A; supplemental Table 2). Twenty-six differentially expressed miRNAs were identified between IM responders and IM nonresponders (P < .05; Figure 1B; supplemental Table 3).

Differentially expressed miRNAs are identified in primary CD34+ CML cells. (A) DESeq2 analysis of differentially expressed miRNAs in CD34+ cells, comparing 3 NBM and 6 CML samples (3 IM responders and 3 IM nonresponders). Plots show the distribution of differentially expressed miRNAs, representing the fold-change between CML relative to NBM (left panel). Red dots represent the differentially expressed miRNAs with adjusted P values < .05. G plots package was used to plot heat maps, accompanied with unsupervised dendrogram analysis, based on the differentially expressed miRNAs with adjusted P values < .05 (right panel). (B) Similar analyses performed for miRNAs found to be differentially expressed between IM responders (R) and IM nonresponders (NR). (C) Differentially expressed miRNAs were determined using a TaqMan qPCR microfluidics device on extracts of CD34+ cells obtained from NBM (n = 11) and CML samples (n = 22). Raw Ct values obtained from the 96-well multiplexing microfluidics device were organized using the HTqPCR package and normalized using the quantile method with the limma package. A nonparametric Mann-Whitney U test was performed to compare unpaired samples. Levels of 20 of the miRNAs tested were shown to be different between CD34+ NBM and CML cells. Data points represent quantile-normalized Ct values relative to CD34+ NBM cells. All comparisons shown are statistically significant (Benjamini-Hochberg-adjusted P value < .05).
Differentially expressed miRNAs are identified in primary CD34+ CML cells. (A) DESeq2 analysis of differentially expressed miRNAs in CD34+ cells, comparing 3 NBM and 6 CML samples (3 IM responders and 3 IM nonresponders). Plots show the distribution of differentially expressed miRNAs, representing the fold-change between CML relative to NBM (left panel). Red dots represent the differentially expressed miRNAs with adjusted P values < .05. G plots package was used to plot heat maps, accompanied with unsupervised dendrogram analysis, based on the differentially expressed miRNAs with adjusted P values < .05 (right panel). (B) Similar analyses performed for miRNAs found to be differentially expressed between IM responders (R) and IM nonresponders (NR). (C) Differentially expressed miRNAs were determined using a TaqMan qPCR microfluidics device on extracts of CD34+ cells obtained from NBM (n = 11) and CML samples (n = 22). Raw Ct values obtained from the 96-well multiplexing microfluidics device were organized using the HTqPCR package and normalized using the quantile method with the limma package. A nonparametric Mann-Whitney U test was performed to compare unpaired samples. Levels of 20 of the miRNAs tested were shown to be different between CD34+ NBM and CML cells. Data points represent quantile-normalized Ct values relative to CD34+ NBM cells. All comparisons shown are statistically significant (Benjamini-Hochberg-adjusted P value < .05).
Sequencing data were validated in CD34+ cells from 11 normal individuals, 11 CML IM responders, and 11 CML IM nonresponders, using a TaqMan qPCR microfluidics device (cohort 1; supplemental Table 1).40 CD34− mononuclear cells from some samples from patients with CML were also included. Twenty miRNAs were confirmed to be differentially expressed in CD34+ CML cells compared with NBM (adjusted P < .05; Figure 1C; supplemental Table 2). Two miRNAs, miR-185 and miR-340, were differentially expressed in CD34+ stem/progenitor cells, but not in CD34− mononuclear cells, when comparing IM responders and IM nonresponders (P = .0006 and .01; Figure 2A; supplemental Table 3). Strikingly, a random forest classifier trained on this data set, with accuracy assessed by the leave-one-out cross-validation method, revealed only miR-185 as a significant classifier to predict TKI response (sensitivity, 90%; specificity, 80%; balance accuracy, 85%; Figure 2B; supplemental Table 4). A logistic regression model also confirmed that CD34+ cells from IM responders and IM nonresponders express significantly different levels of miR-185 (P = .04; supplemental Table 5). We also performed several additional analyses to compare other clinical factors obtained from patients at diagnosis, including age, sex, white blood cell counts, and Sokal scores, which might be associated with CML prognosis and useful for prediction of TKI response, in addition to miRNA expression changes. However, none of these variables proved to be predictive, either alone or in combination with miRNA expression in either a logistic regression or a random forest model (supplemental Tables 6-8). Individually, neither sex nor Sokal score was correlated with TKI response by a χ-square test. Similarly, neither age nor white blood cell count was correlated with TKI response by a Mann-Whitney U test (supplemental Tables 9-10). Interestingly, miR-185’s predictivity of treatment response was further demonstrated in CD34+ cells from an additional 58 treatment-naive patients with CML in a separate cohort, using high-throughput quantitative reverse transcription PCR (qRT-PCR) on the BioMark HD System (Fluidigm; P = .04; Figure 2C). The transcript levels of miR-185 remained significantly lower in nonresponders than responders after patients received therapy with NL for 3 months (P = .014; Figure 2C). Thus, miR-185 was implicated as a new biomarker predictive of TKI responsiveness in primitive, treatment-naive CML patient cells.

Reduced expression of miR-185 predicts therapy response in CD34+ CML cells and miR-185 reexpression restores TKI sensitivity in these cells. (A) TaqMan qRT-PCR was performed to validate differentially expressed miR-185 and miR-340 in CD34+ cells from IM responders (n = 11) and IM nonresponders (n = 11). (B). Random forest classifier analysis of miR-185 and miR-340, assessed by the leave-one-out cross-validation (LOO-CV) method, to predict IM responders and IM nonresponders. (C) Validation of miR-185 transcript levels in CD34+ cells from 47 responders and 11 nonresponders from a second, separate CML cohort before therapy with the TKI nilotinib (baseline) and 3 months posttreatment. (D) Results of CFC assays (± TKIs) of CD34+ CML cells from TKI nonresponders (n = 3) transduced with either a miR-185 expressing vector or a pRRL control vector, with or without 5 µM IM, 150 nM DA, or 5 µM NL treatment. The y-axis shows the frequency of colonies derived from erythroid-burst forming units (BFU-E) and granulocyte/macrophage-colony forming units (CFU-GM; left panel). Results of replating all the cells harvested from the primary CFC assays (left panel) into secondary CFC assays are shown in the right panel. (E) Results of LTC-IC assays performed on the same transduced cells as in panel D, with or without TKIs, as indicated. Data shown are mean ± standard error of the mean (SEM). CFC outputs were measured for cells from 4 individual patients with CML. P values were calculated using a 2-tailed unpaired Student t test.
Reduced expression of miR-185 predicts therapy response in CD34+ CML cells and miR-185 reexpression restores TKI sensitivity in these cells. (A) TaqMan qRT-PCR was performed to validate differentially expressed miR-185 and miR-340 in CD34+ cells from IM responders (n = 11) and IM nonresponders (n = 11). (B). Random forest classifier analysis of miR-185 and miR-340, assessed by the leave-one-out cross-validation (LOO-CV) method, to predict IM responders and IM nonresponders. (C) Validation of miR-185 transcript levels in CD34+ cells from 47 responders and 11 nonresponders from a second, separate CML cohort before therapy with the TKI nilotinib (baseline) and 3 months posttreatment. (D) Results of CFC assays (± TKIs) of CD34+ CML cells from TKI nonresponders (n = 3) transduced with either a miR-185 expressing vector or a pRRL control vector, with or without 5 µM IM, 150 nM DA, or 5 µM NL treatment. The y-axis shows the frequency of colonies derived from erythroid-burst forming units (BFU-E) and granulocyte/macrophage-colony forming units (CFU-GM; left panel). Results of replating all the cells harvested from the primary CFC assays (left panel) into secondary CFC assays are shown in the right panel. (E) Results of LTC-IC assays performed on the same transduced cells as in panel D, with or without TKIs, as indicated. Data shown are mean ± standard error of the mean (SEM). CFC outputs were measured for cells from 4 individual patients with CML. P values were calculated using a 2-tailed unpaired Student t test.
miR-185 functions as a tumor suppressor, and its reexpression in drug-resistant cells restores TKI sensitivity
To investigate the functional significance of differentially expressed miRNAs, we performed a screen of 8 miRNAs by transiently transfecting K562 cells with chemically synthesized miRNA mimics for 7 miRNAs, and an inhibitor for miR-146b (supplemental Figure 2A). Restored expression of 4 miRNAs (miR-145, miR-185, miR-628, and miR-708) moderately reduced K562 cell proliferation (P < .01; supplemental Figure 2B-C). We then conducted biological experiments using an advanced siRNA lentiviral-mediated transduction system in IM-resistant K562 (K562R, a spontaneously derived resistant cell line without BCR-ABL1 mutations),37 parental K562, and BV173 cells (a BCR-ABL1+ALL cell line; supplemental Figure 3).43 miR-185-transduced K562R and BV173 cells displayed reduced growth and increased apoptosis, which were significantly enhanced with IM compared with the lentiviral vector control containing chimeric Rous sarcoma virus (RSV)-HIV 5' LTRs) (pRRL) and other miRNAs (>2-fold; P < .05; supplemental Figures 4 and 5). An IC50 (half maximal inhibitory concentration) assay further showed that miR-185-transduced K562R cells demonstrated increased sensitivity to TKI treatment (IM, DA, NL) in a dose-dependent manner compared with the pRRL vector control (2-6-fold; supplemental Figure 6A).
To determine whether miR-185 contributes to IM sensitization of BCR-ABL1+ cells in a BCR-ABL1 kinase-dependent or BCR-ABL1 kinase-independent manner, miR-185 was stably transduced into UT7-BCR-ABL1+ cells (UT7 B/A) and UT7-BCR-ABL1+ cells carrying a T315I mutant BCR-ABL1 cDNA that prevents IM binding.13 Unlike miR-185-transduced UT7 B/A cells, both UT7-BCR-ABL1-T315I mutant cells and miR-185-transduced UT7 cells (without BCR-ABL1) did not display any effects upon IM, DA, and NL treatments (supplemental Figures 4 and 6B). These results suggest that restored miR-185 expression sensitizes BCR-ABL1+ cells to TKI-induced apoptosis and impairs their proliferation capacity, in part through a BCR-ABL1-kinase-dependent mechanism.
Restored expression of miR-185 significantly impairs survival and increases TKI sensitivity in TKI nonresponder stem/progenitor cells
To investigate the effect of restored expression of miR-185 on primitive cells from patients with CML who do not respond to TKI monotherapy,35 we assessed viability, CFC, and LTC-IC activity (ie, assays for CML progenitor and stem cell functionality).11,12 CD34+ TKI nonresponder cells transduced with the miR-185 expressing vector showed reduced cell growth compared with the same cells transduced with the pRRL control vector, and these effects were significantly enhanced with IM or DA (∼2-fold; P < .05; supplemental Figure 7A). Fewer than 50% of the pretreated CD34+ CML cells from TKI nonresponders responded to TKIs, as reported previously.44 Although the miR-185-transduced cells showed a moderate decrease in total CFCs, these effects were greatly enhanced in replating assays (up to 80% inhibition; P < .01; Figure 3D), suggesting that restored expression of miR-185 sensitizes primitive CML cells to TKIs to an even greater extent. Increased efficacy of the combination on TKI nonresponder LSCs was further demonstrated in LTC-IC assays in which a significant reduction in resistant cells was observed, particularly with DA and NL, the more potent TKIs,5,6 upon reexpression of miR-185 (P < .05; Figure 3E). Importantly, CD34+ normal stem/progenitor cells transduced with the miR-185-expressing vector and treated with TKIs did not demonstrate any inhibitory effect compared with controls (supplemental Figure 7B). Overall, restored expression of miR-185 had a detrimental effect on the survival of patient-derived TKI-insensitive stem/progenitor cells in vitro when exposed to TKIs.
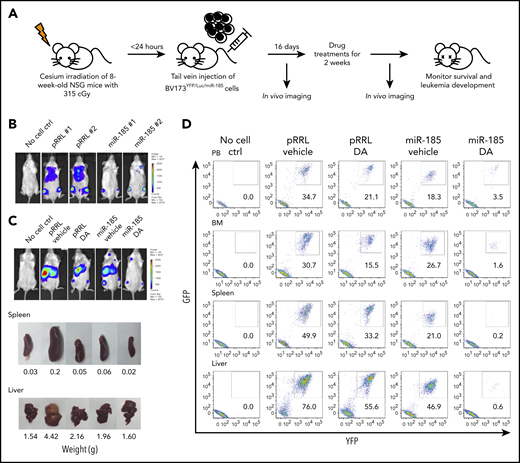

Restoration of miR-185 expression reduces the burden of leukemia and sensitizes leukemic blast cells to TKI treatment, with enhanced survival of leukemic mice. (A) Schematic of the experimental design. BV173YFP/Luc cells transduced with a miR-185-expressing vector, or a pRRL control vector, were injected intravenously into sublethally irradiated nonobese diabetic/severe combined immunodeficiency IL2Rγ-chain–deficient (NSG) mice (2.5 × 106 cells per mouse). Two weeks later, a daily oral gavage treatment of 15 mg/kg DA or vehicle (propylene glycol) was initiated for another 2 weeks. (B) Bioluminescence images of mice taken 2 weeks posttransplantation, before initiation of the oral gavage treatment. (C) Representative bioluminescence images of mice from each group taken 7 weeks posttransplantation. One mouse from each group was then sacrificed to obtain images and weights of spleens and livers. (D) FACS profiles showing human leukemic cell chimerism (GFP+ and YFP+) in PB, BM, spleen, and liver from mice in each group. (E) Representative hematoxylin and eosin-stained sections of spleens and livers from all treatment groups. (F) Fold-difference in BCR-ABL1 transcripts in hematopoietic tissues from each group. Data shown are mean ± SEM of measurements from 3 technical replicates. P values were calculated using a 2-tailed paired Student t test. (G) Western blots of whole protein extracted from BM cells of mice in each group, after probing with the antibodies indicated. (H) Overall survival of mice from each treatment group (n = 6 mice per group). P values were calculated using log-rank test. ND = not detectable.
Restoration of miR-185 expression reduces the burden of leukemia and sensitizes leukemic blast cells to TKI treatment, with enhanced survival of leukemic mice. (A) Schematic of the experimental design. BV173YFP/Luc cells transduced with a miR-185-expressing vector, or a pRRL control vector, were injected intravenously into sublethally irradiated nonobese diabetic/severe combined immunodeficiency IL2Rγ-chain–deficient (NSG) mice (2.5 × 106 cells per mouse). Two weeks later, a daily oral gavage treatment of 15 mg/kg DA or vehicle (propylene glycol) was initiated for another 2 weeks. (B) Bioluminescence images of mice taken 2 weeks posttransplantation, before initiation of the oral gavage treatment. (C) Representative bioluminescence images of mice from each group taken 7 weeks posttransplantation. One mouse from each group was then sacrificed to obtain images and weights of spleens and livers. (D) FACS profiles showing human leukemic cell chimerism (GFP+ and YFP+) in PB, BM, spleen, and liver from mice in each group. (E) Representative hematoxylin and eosin-stained sections of spleens and livers from all treatment groups. (F) Fold-difference in BCR-ABL1 transcripts in hematopoietic tissues from each group. Data shown are mean ± SEM of measurements from 3 technical replicates. P values were calculated using a 2-tailed paired Student t test. (G) Western blots of whole protein extracted from BM cells of mice in each group, after probing with the antibodies indicated. (H) Overall survival of mice from each treatment group (n = 6 mice per group). P values were calculated using log-rank test. ND = not detectable.
Restored miR-185 expression markedly sensitizes leukemic blast cells to TKI treatment and enhances survival of leukemic mice
To determine whether restored expression of miR-185, particularly with TKIs, can effectively target aggressive BCR-ABL1+ blast cells from late-stage disease where TKI monotherapy is ineffective,15,16 we examined its effect on BV173 cells, which generate a lethal leukemia in transplanted immunodeficient mice (Figure 3A).42 Control, pRRL vector-transduced, or miR-185-tranduced BV173YFP+/Luc cells, carrying a luciferase reporter, were intravenously injected into sublethally irradiated nonobese diabetic/severe combined immunodeficiency IL2Rγ–deficient mice. Bioluminescence imaging demonstrated that after only 2 weeks, mice injected with miR-185-tranduced BV173YFP/Luc cells had greatly reduced chimerism compared with controls (Figure 3B). Mice were then treated with 15 mg/kg DA or propylene glycol (the vehicle control) for 2 weeks by oral gavage and assessed again another 3 weeks later. Within 7 to 8 weeks, vehicle control-treated recipients of pRRL-transduced BV173 cells developed an aggressive widely distributed leukemia, with enlarged spleens and livers (0.2 and 4.4 g, respectively), and even recipients of pRRL-transduced cells treated with DA, or vehicle control-treated mice originally injected with miR-185-transduced cells, displayed slight splenomegaly (0.05 and 0.06 g, respectively) and hepatomegaly (2.2 and 1.9 g, respectively; Figure 3C-D). However, mice injected with miR-185-transduced cells and then given DA had no leukemia progression, with dramatically lower chimerism in the BM (1.6%), spleen (0.2%), and liver (0.6%), and without evidence of spleen (0.02 g) or liver (1.6 g) enlargements (Figure 3C-E). qRT-PCR and western blot analyses confirmed a significant reduction in leukemia burden, with decreased BCR-ABL1 transcript levels, and phosphorylation and protein expression of BCR-ABL1, p-STAT5, and p-CRKL in mice injected with miR-185-transduced cells and treated with DA (Figure 3F-G).
These results were supported by analysis 6 weeks after discontinuation of treatment (10 weeks posttransplant; supplemental Figure 8). Overall, we observed significantly longer survival in mice injected with miR-185-transduced cells vs pRRL control cells (6 mice per group, median survival, 65 vs 47.5 days; P = .0005). Strikingly, mice injected with miR-185-transduced cells and treated with DA survived much longer compared with recipients of pRRL-transduced cells treated with DA (median survival, 83 vs 60 days; P = .0018; Figure 3H). Therefore, restoration of miR-185 expression can reduce aggressively advancing BCR-ABL1+ leukemia in vivo, and this effect can be greatly enhanced by DA treatment, resulting in effective leukemia elimination and a significant survival advantage.
Restoration of miR-185, combined with TKI treatment, eliminates initiation of CML in mice transplanted with IM nonresponder stem/progenitor cells
To investigate whether restored miR-185 expression affects survival of long-term leukemia initiating LSCs, we transduced CD34+ CML cells from 2 TKI nonresponder patients with pRRL or miR-185 vectors carrying a GFP marker, and injected these cells either directly or after exposure to DA overnight into sublethally irradiated immunocompromised nonobese diabetic-Rag1−/−IL2Rγc−/− mice (supplemental Figure 9A).45 Four weeks later, there was a markedly lower level of human GFP+ cells in the PB and BM of recipients of miR-185-transduced cells compared with pRRL-transduced control cells, and this effect was significantly enhanced on DA treatment in both experiments (P < .04; Figure 4A; supplemental Figure 9F). This decrease was shared by GFP+ myeloid (CD33+/15+), CD34+, and CD34+CD38− LSCs in the recipients of the miR-185-transduced cells treated with DA (supplemental Figure 9B-E). Despite an overall lower level of chimerism at week 12, similar differences were sustained as in week 4 (P = .032; Figure 4A; supplemental Figure 9C-E). At 25 weeks posttransplantation, the mice had very few miR-185-transduced and DA-treated GFP+ leukemic cells in the PB or BM (Figure 4A,C; supplemental Figure 9G). In particular, we observed not only a marked reduction in GFP+CD34+ cells but also a near elimination of GFP+CD34+CD38− LSCs that had been transduced with miR-185 and treated with DA compared with cells transduced with the pRRL vector and treated with DA (Figure 4D-E). There were also no detectable CFCs in assays of FACS-sorted GFP+ BM cells at 25 weeks posttransplant and very low levels of BCR-ABL1 transcripts in mice injected with miR185-transduced cells that received DA treatment compared with other groups in both experiments (Figure 4B,F). Overall, restored miR-185 expression combined with DA treatment preferentially prevents the growth of patient-derived long-term leukemia-initiating cells in a patient-derived xenotransplantation model.

Restoration of miR-185 expression eliminates TKI nonresponder cells regenerated in transplanted nonobese diabetic-Rag1−/−IL2Rγc−/− mice. (A) Detection of human GFP+CD45+ cells in the BM of mice injected with CD34+ CML patient cells transduced with a pRRL control vector (light blue), a miR-185 vector (light red), pRRL plus DA treatment (dark blue), or miR-185 plus DA treatment (dark red) at weeks 4, 12, and 25 posttransplantation. (B) qRT-PCR results for BCR-ABL1 transcript levels relative to GAPDH in total BM cells of all mice analyzed at 25 weeks. (C) Representative FACS plots of human (CD45+) and GFP+CD45+ cells detected in the BM of mice analyzed at 25 weeks posttransplantation (left) and a summary of the levels of transduced GFP+CD45+ (solid bars) versus non-transduced GFP-CD45+ (white bars, right) human cells in all mice studied. (D) Levels of human GFP+CD34+, CD34+CD38−, and myeloid cells detected in the BM of all mice analyzed at week 25 posttransplantation. (E) Representative FACS plots of human GFP+CD34+ cells in the BM of mice at week 25 posttransplantation. (F) Colony output of FACS-sorted human GFP+CD45+ cells harvested from the BM of mice 25 weeks posttransplantation of cells from a CML patient sample. Data shown, except where otherwise indicated, are the mean ± SEM of measurements from 2 cohorts of mice injected with transduced cells from 2 independent TKI nonresponder patients. Each dot/triangle represents 1 mouse. P values were calculated using a 2-tailed unpaired Student t test. No. = number.
Restoration of miR-185 expression eliminates TKI nonresponder cells regenerated in transplanted nonobese diabetic-Rag1−/−IL2Rγc−/− mice. (A) Detection of human GFP+CD45+ cells in the BM of mice injected with CD34+ CML patient cells transduced with a pRRL control vector (light blue), a miR-185 vector (light red), pRRL plus DA treatment (dark blue), or miR-185 plus DA treatment (dark red) at weeks 4, 12, and 25 posttransplantation. (B) qRT-PCR results for BCR-ABL1 transcript levels relative to GAPDH in total BM cells of all mice analyzed at 25 weeks. (C) Representative FACS plots of human (CD45+) and GFP+CD45+ cells detected in the BM of mice analyzed at 25 weeks posttransplantation (left) and a summary of the levels of transduced GFP+CD45+ (solid bars) versus non-transduced GFP-CD45+ (white bars, right) human cells in all mice studied. (D) Levels of human GFP+CD34+, CD34+CD38−, and myeloid cells detected in the BM of all mice analyzed at week 25 posttransplantation. (E) Representative FACS plots of human GFP+CD34+ cells in the BM of mice at week 25 posttransplantation. (F) Colony output of FACS-sorted human GFP+CD45+ cells harvested from the BM of mice 25 weeks posttransplantation of cells from a CML patient sample. Data shown, except where otherwise indicated, are the mean ± SEM of measurements from 2 cohorts of mice injected with transduced cells from 2 independent TKI nonresponder patients. Each dot/triangle represents 1 mouse. P values were calculated using a 2-tailed unpaired Student t test. No. = number.
RNA sequencing analysis uncovers perturbations of oxidative phosphorylation and reactive oxygen species in TKI nonresponder cells and identifies PAK6 as a critical target of miR-185
We next used strand-specific RNA sequencing of the same 9 RNA samples to correlate miRNA profiles with corresponding gene expression changes. Of the 21 727 protein-coding genes sequenced, more than 6000 were differentially expressed in CD34+ CML cells compared with their normal BM counterparts (P < .05). Of these, 547 were downregulated and 560 were upregulated when IM nonresponders and IM responders were compared (P < .05; Figure 5A). Interestingly, gene set enrichment analysis identified a significant gene set enrichment of mitochondrial oxidative phosphorylation (OXPHOS) and reactive oxygen species (ROS) in CD34+ CML cells compared with healthy CD34+ cells (nominal enrichment scores, 2.44 and 1.65, respectively; Figure 5B). Moreover, these changes were significantly greater in nonresponders than responders (nominal enrichment scores, 1.73 and 0.49, respectively; Figure 5B). These results indicate that metabolic pathways are perturbed in primitive CML cells, particularly in TKI nonresponder LSC/progenitor cells, and may confer therapy resistance.


RNA-seq analysis identifies increased expression of OXPHOS and ROS gene signatures in TKI nonresponder cells and PAK6 as a target of miR-185. (A) DESeq2 analysis of differentially expressed genes from the same 9 RNA samples used for miRNA profiling and a comparison of genes expressed differentially in CD34+ cells from NBM (n = 3) vs CML (n = 6, top panel) samples and IM responder (n = 3) vs IM nonresponder (n = 3, bottom panel) samples. Red dots represent differentially expressed genes with adjusted P values < .05 between these samples. (B) Gene set enrichment analysis plots for OXPHOS and ROS pathways, with nominal enrichment scores. (C) Heat maps of miR-185-predicted target genes identified in CD34+ cells from NBM vs CML cells. (D) qRT-PCR analysis of miR-185 expression in IM-resistant K562R cells, CD34+ IM responder (n = 3) and IM nonresponder (n = 3) cells transduced with a miR-185-expressing vector or a pRRL control vector and incubated with or without IM (5.0 µM) for 24 hours. (E) Western blot analysis of several key signaling proteins in miR-185-transduced K562R cells, PAK6 siRNA-transfected K562R cells, and control cells cultured with or without IM (5.0 µM) for 24 hours. ACTIN served as loading control. (F-H) qRT-PCR analyses of the transcript levels of PAK6 in CD34+ cells from NBM vs IM responders (R) or IM nonresponders (NR). Correlation analysis of PAK6 and miR-185 expression in responder and nonresponder patients (G), and PAK6 transcript levels in CD34 subpopulations from responders vs nonresponders. P values were calculated using a 2-tailed paired (Figure 6F) or unpaired Student t test. Ctrl = control.
RNA-seq analysis identifies increased expression of OXPHOS and ROS gene signatures in TKI nonresponder cells and PAK6 as a target of miR-185. (A) DESeq2 analysis of differentially expressed genes from the same 9 RNA samples used for miRNA profiling and a comparison of genes expressed differentially in CD34+ cells from NBM (n = 3) vs CML (n = 6, top panel) samples and IM responder (n = 3) vs IM nonresponder (n = 3, bottom panel) samples. Red dots represent differentially expressed genes with adjusted P values < .05 between these samples. (B) Gene set enrichment analysis plots for OXPHOS and ROS pathways, with nominal enrichment scores. (C) Heat maps of miR-185-predicted target genes identified in CD34+ cells from NBM vs CML cells. (D) qRT-PCR analysis of miR-185 expression in IM-resistant K562R cells, CD34+ IM responder (n = 3) and IM nonresponder (n = 3) cells transduced with a miR-185-expressing vector or a pRRL control vector and incubated with or without IM (5.0 µM) for 24 hours. (E) Western blot analysis of several key signaling proteins in miR-185-transduced K562R cells, PAK6 siRNA-transfected K562R cells, and control cells cultured with or without IM (5.0 µM) for 24 hours. ACTIN served as loading control. (F-H) qRT-PCR analyses of the transcript levels of PAK6 in CD34+ cells from NBM vs IM responders (R) or IM nonresponders (NR). Correlation analysis of PAK6 and miR-185 expression in responder and nonresponder patients (G), and PAK6 transcript levels in CD34 subpopulations from responders vs nonresponders. P values were calculated using a 2-tailed paired (Figure 6F) or unpaired Student t test. Ctrl = control.
To further identify target genes, several prediction algorithms were applied to match the differentially expressed genes to the 20 deregulated miRNAs that were identified by miRNA profiling, with an inversely correlated expression profile (Figures 1 and 5A; supplemental Tables 11 and 12; supplemental Figure 10). Several of the predicted miR-185 target genes identified showed significantly increased expression in CD34+ CML cells compared with normal BM cells, or in IM nonresponders compared with IM responders (P < .05; Figure 5C). Four of these, PBX1, PAK6, SGMS1, and NR1D1, were selected for further study based on their highly deregulated expression in CML stem/progenitor cells and potential biological significance.46-49 PAK6 was confirmed to be the most relevant bona fide target gene of miR-185 by a luciferase reporter assay (P < .05; supplemental Figure 11A-B), and a marked decrease in PAK6 transcripts and protein was demonstrated when miR-185 expression was restored in IM-resistant cells compared with control cells and other target genes (P < .05; supplemental Figures 11D and 12A). These results were further supported by knockdown of PAK6 in IM-resistant cells, which displayed significantly increased sensitivity to IM (P < .01; supplemental Figures 11E and 12B). Thus, PAK6 is a novel target of miR-185, and loss of miR-185 expression in CML cells appears to lead to upregulated expression of PAK6 and decreased sensitivity to TKIs.
Increased expression of PAK6 in TKI nonresponder cells is correlated with downregulation of miR-185, and targeting miR-185-mediated PAK6 activity sensitizes these cells to TKIs
We next investigated several potential pathways that might be affected by miR-185-regulated PAK6 activity in IM nonresponder cells. IM treatment of miR-185-transduced K562R cells and CD34+ IM nonresponder cells (but not CD34+ IM responder cells) revealed increased miR-185 expression, suggesting that it might be regulated by BCR-ABL1 (Figure 5D). Western blot analysis also revealed that IM treatment of miR-185-transduced K562R cells caused a reduction in p-BCR-ABL1 and p-ERK compared with control vector transduced cells. Notably, reduced p-ERK and p-AKT were more pronounced in PAK6 knockdown cells in response to IM treatment (Figure 5E). These results suggest that miR-185 was upregulated by IM, thereby inducing these cells to undergo TKI-induced apoptosis through inhibition of the RAS/MAPK pathway, mediated by PAK6 (Figure 6F). Thus, we hypothesized that inhibition of PAK6 activity by upregulating miR-185 may directly reverse the insensitivity of TKI-resistant cells (in which PAK6 is increased). Indeed, PAK6 transcripts were highly increased in CD34+ CML cells obtained from patients with CML at diagnosis before IM therapy (cohort 1, n = 24) compared with healthy CD34+ cells (n = 11; P = .0001; Figure 5F). In particular, PAK6 was highly elevated in IM nonresponder cells, including LSCs, compared with IM responders (P = .0038; Figure 5F,H), and its expression correlated significantly with downregulation of miR-185 expression (P = .002; Figure 5G). We then investigated whether PAK6 inhibition could modulate changes in OXPHOS and ROS production in IM nonresponder cells. Interestingly, MitoTracker analysis showed that a preclinically validated pan-PAK inhibitor (PF-3758309) alone,50 or in combination with a TKI, significantly reduced mitochondrial activity in IM nonresponder cells, an effect not seen with TKI alone (P < .002; Figure 6A). Notably, ROS production was also greatly reduced in these cells treated with PF-3758309 (Figure 6B). Most interestingly, PF-3758309 significantly reduced the growth of K562R, BV173 blast cells (IC50, 25-70 nM), and CD34+ IM nonresponder cells, as assessed by viability and CFC assays, and increased their apoptosis; these effects were greatly enhanced by TKIs (2-fold; P < .05; Figure 6C-E; supplemental Figure 12C). In addition, PF-3758309 alone, or in combination with a TKI, did not have an obvious inhibitory effect on CD34+ normal BM cells (supplemental Figure 12D). Thus, PAK6, a novel target of miR-185, emerges as an attractive druggable target for combination therapy of TKI-resistant patients.
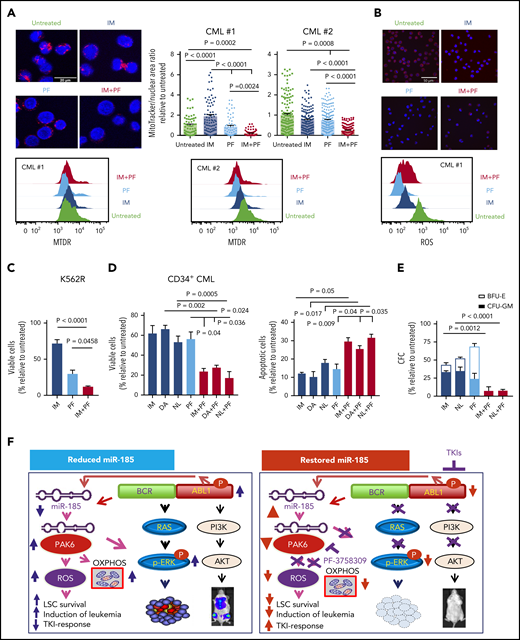
The PAK inhibitor PF-3758309 sensitizes CD34+ nonresponder cells to TKIs and perturbs mitochondrial activity. (A) Representative confocal images showing the morphology of mitochondria stained in CD34+ IM nonresponder cells treated with PF-3758309 (PF) and IM, as compared with control and single drug treatment and quantification of mitochondria to nuclear area ratio in these treated cells are shown and each dot represents an individual cell (top, n = 2). The white scale bar represents 20 μm. Intracellular mitochondrial staining of the same cells by MitoTracker Deep Red (MTDR) is also shown (bottom). (B) ROS was assessed using CellROX Deep Red in the same cells as in (A), and representative images and intracellular ROS accumulation are shown. The white scale bar represents 50 μm. (C-D) Viability and apoptosis assays of IM-resistant K562 cells (K562R) and CD34+ cells from IM nonresponders (n = 3) for 72 hours in the presence or absence of PF or TKIs (IM, DA, and NL), alone or in combination. (E) CFC assays of CD34+ CML cells from TKI nonresponders (n = 3) with or without PF or TKIs, alone or in combination. Data shown are mean ± SEM of measurements from 3 patient samples. P values were calculated using a 2-tailed unpaired Student t test. (F) Model of how restored expression of miR-185 or inhibition of PAK6 activity sensitizes TKI-resistant CML cells to TKIs both in vitro and in vivo, by dual targeting of a novel miR-185-PAK6 axis and BCR-ABL1 activity.
The PAK inhibitor PF-3758309 sensitizes CD34+ nonresponder cells to TKIs and perturbs mitochondrial activity. (A) Representative confocal images showing the morphology of mitochondria stained in CD34+ IM nonresponder cells treated with PF-3758309 (PF) and IM, as compared with control and single drug treatment and quantification of mitochondria to nuclear area ratio in these treated cells are shown and each dot represents an individual cell (top, n = 2). The white scale bar represents 20 μm. Intracellular mitochondrial staining of the same cells by MitoTracker Deep Red (MTDR) is also shown (bottom). (B) ROS was assessed using CellROX Deep Red in the same cells as in (A), and representative images and intracellular ROS accumulation are shown. The white scale bar represents 50 μm. (C-D) Viability and apoptosis assays of IM-resistant K562 cells (K562R) and CD34+ cells from IM nonresponders (n = 3) for 72 hours in the presence or absence of PF or TKIs (IM, DA, and NL), alone or in combination. (E) CFC assays of CD34+ CML cells from TKI nonresponders (n = 3) with or without PF or TKIs, alone or in combination. Data shown are mean ± SEM of measurements from 3 patient samples. P values were calculated using a 2-tailed unpaired Student t test. (F) Model of how restored expression of miR-185 or inhibition of PAK6 activity sensitizes TKI-resistant CML cells to TKIs both in vitro and in vivo, by dual targeting of a novel miR-185-PAK6 axis and BCR-ABL1 activity.
Discussion
Subsets of malignant cells play important roles in disease progression and recurrence because of their intrinsic ability for self-propagation and insensitivity to available treatments.1,3,12 This phenomenon has been most intensively studied in human leukemias, including CML, where targeted drugs have improved outcomes, but resistance remains problematic. Our study now identifies miR-185, which is downregulated in the responsible cells and governs their sensitivity and resistance to drugs that target the key oncoprotein that drives the disease process.1,3-5,13 The fact that levels of miR-185 correlated with TKI sensitivity not only in patients at diagnosis but also in patients after receiving 3 months of therapy points to the potential value of its measurement as a guide to treatment, by rapid early identification of patients with CML who are unlikely to respond to TKI monotherapies. In addition, functional studies demonstrated that restoration of miR-185 expression in TKI nonresponder cells, including BCR-ABL1+ ALL cells, significantly impairs their survival and sensitizes them to TKI treatment in vitro and in vivo in a fashion that may be enhanced by PAK6 inhibition.
miR-185 was previously found to be downregulated and to affect tumor growth in several types of solid tumors,51,52 but an ability to control the properties of cells that propagate malignant populations, including their drug resistance, has not been previously appreciated, and its target genes have remained largely unknown. Here we provide the first evidence that miR-185 has clinically predictive and therapeutic value in human leukemia. Interestingly, we observed that miR-185 expression is increased in both miR-185-transduced K562-resistant cells and CD34+ IM nonresponder cells in response to IM treatment, but did not observe obvious changes in miR-185-transduced IM responder cells, suggesting that its downregulation in IM nonresponder cells is meditated in part by BCR-ABL1 kinase. It is thus interesting to note that BCR-ABL1 transcripts and protein are significantly elevated in CD34+, and even more so in the CD34+CD38− LSCs of CML cells compared with the bulk CD34− population.12,53 It is also the CD34+ stem and progenitor cells that are the least TKI responsive3,10-12 and are responsible for disease recurrence when TKIs are stopped.8,9
We further demonstrated that miR-185 activates resistance to TKIs via a mechanism that may have broader relevance in human cancers, given its ability to target PAK6 transcripts. The PAK6 gene encodes a member of a family of p21-stimulated serine/threonine protein kinases that contain an amino-terminal Cdc42/Rac interactive binding domain and a carboxyl-terminal kinase domain. The PAK6 protein affects multiple cell processes, including the cytoskeleton and transcription. Notably, expression of some PAK family members has been correlated with poor outcomes in various solid tumors, and PAK6, specifically, plays a role in the development of prostate cancer and acquisition of chemoresistance in colon cancer.46,54-56 In CML, it appears that reactivated miR-185 expression may promote TKI-dependent apoptosis by suppressing PAK6 activity, and hence the MAPK pathway. Consistent with this mechanism is a report in which TKI treatment of BCR-ABL1+ cells leads to activation of the MAPK/ERK pathway and a combination of TKI and MEK inhibitors induces synthetic lethality in IM-resistant cells.57 Notably, additional targets of miR-185 were also identified in this study, including PBX1, SGMS1, and NR1D147-49 ; their expression was deregulated in CML stem/progenitor cells, potentially contributing to TKI resistance. Thus their specific biological significance remains to be determined.
Importantly, we have provided new evidence that mitochondrial OXPHOS and ROS pathways are enriched in TKI nonresponder stem/progenitor cells and that pharmacological inhibition of PAK6 reduces mitochondrial activity and ROS production and inhibits the growth of TKI nonresponder cells; this is significantly enhanced by TKIs. These findings point to PAK6-mediated oxidative metabolism as a critical survival mechanism of drug-resistant LSC/progenitor cells from TKI nonresponders. Recently, there has been renewed interest in targeting cancer metabolism/mitochondria for anticancer therapies. Evidence indicates that cancer stem cells are susceptible to cellular metabolic changes and seem to have a greater dependence on OXPHOS for survival, which may constitute a targetable vulnerability.58-61 It has also been reported that deregulated ROS production causes genomic instability and affects autophagy, leading to TKI resistance and disease progression.62,63 Thus, identification of a highly effective approach for treating TKI nonresponder BCR-ABL1+ cells with leukemia initiating capacity in vivo, using available drugs, opens up a new avenue for developing improved treatment options for curative treatment of CML, including a subset of currently fatal leukemias.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the Stem Cell Assay Laboratory staff for processing patient samples, Josephine Leung and Glenn Edin for excellent technical assistance, members of the Leukemia/Bone Marrow Transplant Program of British Columbia and the Hematology Cell Bank of British Columbia for patient samples, the Terry Fox Laboratory FACS Facility for cell sorting, STEMCELL Technologies for culture reagents, and A. Turhan for IM-resistant cell lines.
This work was supported by the Canadian Cancer Society (Grant #705148), the Leukemia & Lymphoma Society of Canada and the Canadian Institutes of Health Research (CIHR, Grant #427201 to X.J.), and the Cancer Research Society (C.J.E.). H.L. received a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship; K.R. is the recipient of a Mathematics of Information Technology and Complex Systems (MITACS) Elevate postdoctoral fellowship; A.B. is recipient of the Roman M. Babicki Fellowship in Medical Research from the University of British Columbia (UBC); and R.Y. is recipient of a Four-Year Fellowship from UBC and a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship.
Authorship
Contribution: H.L. and K.R. designed and performed most experiments and analyzed data; H.L, K.R., C.J.E., and X.J. wrote the manuscript and all authors commented on it; M.C., A.W., R.Y., and J.Z. conducted some experiments and analyzed data; J.R. and O.I.P. helped to perform TaqMan qPCR microfluidics experiments; A.B., K.O., and D.J.H.F.K. performed statistical and bioinformatics analyses; R.B. and I.B. provided oversight of statistical and bioinformatics analyses; T.M. and R.K.H. designed a lentiviral vector to transduce primary CML cells; D.L.F. provided clinical data; C.H. and R.K.H. provided expertise in TaqMan qPCR microfluidics analysis; N.N. and C.J.E. provided expertise in xenotransplantation models; and X.J. developed the concept, designed the experiments, and supervised the study.
Conflict-of-interest disclosure: D.L.F., C.J.E., and X.J., received funding from Novartis Canada; X.J. received funding from Bristol-Myers Squibb and Pfizer. The remaining authors declare no competing financial interests.
Correspondence: Xiaoyan Jiang, Terry Fox Laboratory, BC Cancer Research Institute, 675 West 10th Ave, Vancouver, BC V5Z 1L3, Canada; e-mail: xjiang@bccrc.ca.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal