

Key Points
Trib1 affects Hoxa9 in myeloid leukemogenesis via degradation of C/EBPα and modification of super-enhancers associated with Hoxa9.
Erg, a critical target of Trib1 and Hoxa9, is responsive to BRD4 inhibition and plays a key role in leukemogenesis.

Abstract
The pseudokinase Trib1 functions as a myeloid oncogene that recruits the E3 ubiquitin ligase COP1 to C/EBPα and interacts with MEK1 to enhance extracellular signal-regulated kinase (ERK) phosphorylation. A close genetic effect of Trib1 on Hoxa9 has been observed in myeloid leukemogenesis, where Trib1 overexpression significantly accelerates Hoxa9-induced leukemia onset. However, the mechanism underlying how Trib1 functionally modulates Hoxa9 transcription activity is unclear. Herein, we provide evidence that Trib1 modulates Hoxa9-associated super-enhancers. Chromatin immunoprecipitation sequencing analysis identified increased histone H3K27Ac signals at super-enhancers of the Erg, Spns2, Rgl1, and Pik3cd loci, as well as increased messenger RNA expression of these genes. Modification of super-enhancer activity was mostly achieved via the degradation of C/EBPα p42 by Trib1, with a slight contribution from the MEK/ERK pathway. Silencing of Erg abrogated the growth advantage acquired by Trib1 overexpression, indicating that Erg is a critical downstream target of the Trib1/Hoxa9 axis. Moreover, treatment of acute myeloid leukemia (AML) cells with the BRD4 inhibitor JQ1 showed growth inhibition in a Trib1/Erg-dependent manner both in vitro and in vivo. Upregulation of ERG by TRIB1 was also observed in human AML cell lines, suggesting that Trib1 is a potential therapeutic target of Hoxa9-associated AML. Taken together, our study demonstrates a novel mechanism by which Trib1 modulates chromatin and Hoxa9-driven transcription in myeloid leukemogenesis.
Introduction
Aberrations of transcriptional regulation are common molecular characteristics of acute myeloid leukemia (AML). Hematopoietic transcription factors have been frequently identified as oncogenes or tumor suppressor genes in AML. Hox proteins are key transcription factors, and the transcriptional program regulated by Hoxa9 is involved in AML with MLL fusions, MOZ-TIF2, NUP98-NSD1, NPM1c, or ASXL1 fusions/mutations.1,2 These mutations upregulate the messenger RNA (mRNA) expression of HOXA9, and the significance of Hoxa9 overexpression has been revealed to immortalize murine hematopoietic cells.3 However, simple overexpression of Hoxa9 is not sufficient for developing full-blown AML, as activation of additional cofactors and/or cooperative pathways is required.
Hoxa9 and other abdB-type Hoxa genes are critical transcription factors regulating hematopoiesis and leukemogenesis.4 Overexpression of HOXA9 is associated with poor prognosis in normal-karyotype AML,1 and HOXA9 is a critical direct downstream target of MLL fusion proteins.5-7 Hoxa9 overexpression is necessary for the immortalization of murine hematopoietic cells; however, it is insufficient for developing in vivo leukemogenesis, as interactions with specific cofactors such as Meis1 or its downstream target, Sytl1, are needed.3 Sytl1 facilitates membrane trafficking of cytoplasmic vesicles that contain CXCR4, resulting in the promotion of leukemic cell engraftment in bone marrow. The importance of leukemic cell engraftment via the Meis1/Sytl1 axis indicates that transcriptional programs regulated by Hoxa9 do not sufficiently support leukemic cell engraftment and/or interaction with bone marrow stroma. Activation of the Syk protein kinase was also found to interact with Hoxa9 by activating Meis1 via suppressing miR-146a.8 Syk-mediated signaling also induces PU.1 suppression via Meis1, suggesting that modifications of multiple signaling pathways may promote expansion of leukemia cell transformed by Hoxa9 in vivo. These studies suggest that multiple signaling pathways interact with the transcriptional program regulated by Hoxa9 and that additional molecular mechanisms may affect the Hoxa9-driven enhancer modifications.
The pseudokinase Trib1 plays an important role in differentiation and neoplastic transformation in the myeloid lineage compartment.9-12 Trib1 interacts with the E3 ubiquitin ligase Cop1 to degrade the C/EBPα transcription factor and with MEK1 to facilitate phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2).13,14 Degradation of C/EBPα p42 and enhancement of MEK/ERK signaling contribute to the strong driving force of Trib1 behind myeloid leukemogenesis.10,14 Both Trib1 and Trib2 also have an effect on Hoxa9, a key factor in AML development and malignant progression.2,9,14,15 The genetic interaction between Trib1 and Hoxa9 is specific9 ; however, the mechanisms behind this interaction remain to be clarified. Several studies showed that the myeloid transcription factor C/EBPα and Hoxa9 frequently share transcriptional targets, as they cobind identical loci.16,17 In this regard, it is possible that C/EBPα p42 degradation by Trib1 induces dynamic enhancer remodeling in the Hoxa9-associated genomic loci.
To reveal the role of Trib1 in the promotion and progression of Hoxa9-induced leukemogenesis, we aimed to examine whether Trib1 modulates enhancer programs of Hoxa9 in association with C/EBPα. Trib1 overexpression induced super-enhancers at certain target loci such as Erg and Spns2. The BRD4 inhibitor JQ1 inhibited super-enhancer signals and expression of target genes, resulting in growth inhibition of Trib1-expressing AML both in vitro and in vivo. Moreover, histone H3K27Ac signals of the super-enhancers and expression of the target genes were suppressed by the JQ1 treatment in a Trib1-dependent manner. Our study delivers new insights into the Trib1 function and its role in myeloid leukemogenesis and provides potential novel therapeutic strategies.
Methods
Mice and bone marrow transplantation
Trib1 conditional knockout mice were generated by inserting loxP sequences in Trib1 intron 1 and 325 bp downstream of exon 3. The mice were mated with ROSA26-Cre deleter mice, and a germline knockout allele was created. For transplantations, mice were subjected to 4.0-Gy irradiation and injected IV with 1 × 107 Trib1 hi or null cells. All mice were on a C57Bl/6 background, and wild-type mice were purchased from Clea Japan. A detailed protocol of in vivo drug treatment is described in supplemental Methods (available on the Blood Web site). All experiments described in this study were performed in strict accordance with standard ethical guidelines and approved by the animal care committee at Japanese Foundation for Cancer Research under licenses 10-05-9 and 0604-3-13. This study was conducted in accordance with the Declaration of Helsinki.
Plasmids
Hoxa9, Trib1, and Erg complementary DNAs (cDNAs) were cloned into pMYs retroviral vectors. Lentivirus plasmids containing short hairpin RNA (shRNA) constructs of mouse Erg, Cebpa, human TRIB1, and non-target control were purchased from Sigma-Aldrich (St. Louis, MO). A lentivirus plasmid containing short guided RNA (sgRNA) for Erg was purchased from Addgene (Watertown, MA). The shRNA and sgRNA sequences are listed in supplemental Table 1.
Cell lines and cell culture
Bone marrow cells were prepared from an 8-week-old female Trib1ko/ko mouse 5 days after injecting 150 mg of 5-fluorouracil/kg body weight (Kyowa Hakko Kirin, Tokyo, Japan). Bone marrow cells were transduced with pMYs-Hoxa9-IRES-mKO with or without pMYs-FLAG-Trib1-IRES-EGFP as previously described.3 Trib1 hi and null cells were maintained in Iscove modified Dulbecco medium containing 10% fetal bovine serum and 5 ng/mL interleukin-3. Human HEL, KU812, P39, HL-60, and THP1 AML cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum.
Immunoblotting
Western blot analysis was performed as previously described18 using whole-cell lysates and the specific antibodies listed in supplemental Table 2.
Flow cytometry
Cells were analyzed on a FACSAria flow cytometer (BD Biosciences, Franklin Lakes, NJ) and data were analyzed using FlowJo software v.10.5 (TreeStar, Ashland, OR). The antibodies used are listed in supplemental Table 2.
Gene expression profiling
ChIP-seq
Chromatin immunoprecipitation sequencing (ChIP-seq) was performed as previously described,20 with modifications. A total of 5 × 107 AML cells per immunoprecipitation reaction were cross-linked with 1% formaldehyde for 10 minutes at room temperature. ChIP was performed with 4 μg anti-histone H3K27ac (Active Motif, Carlsbad, CA), anti-FLAG (Sigma-Aldrich), or anti-C/EBPα (Santa Cruz Biotechnology, Dallas, TX) antibodies. The results were visualized using IGV_2.3.80 (http://software.broadinstitute.org/software/igv). Hoxa9- and C/EBPα-binding motifs were searched using the AME (MEME-Suit version 4.11.2) program (http://meme-suite.org/index.html). Super-enhancers were identified as previously described21,22 with the ROSE program (http://younglab.wi.mit.edu/super_enhancer_code.html). Gene Ontology analyses for super-enhancers were performed using GREAT version 4.0.4 (http://great.stanford.edu/public/html/).23 Sequences of primers used for ChIP-quantitative polymerase chain reaction (PCR) are listed in supplemental Table 3.
Statistical analysis
All in vitro experiments were performed at least in triplicate. The numbers of mice used per experiment is indicated in the text or figure legends. Values are expressed as mean ± SD, and statistical significance was determined using a 2-tailed Student t test. Survival analysis was performed using the Kaplan-Meier life table method, and survival between groups was compared with the log-rank test.
Results
Trib1 overexpression promotes Hoxa9-induced myeloid leukemogenesis
Our previous study indicated that Trib1 overexpression accelerated Hoxa9-induced myeloid leukemogenesis9 ; however, the mechanistic basis underlying the effect Trib1 has on Hoxa9 interaction remained unclear. To clarify the biological role of Trib1 in Hoxa9-expressing leukemic cells, immortalized bone marrow cell lines were established by introducing Hoxa9 and/or Trib1 using the retrovirus vector pMYs-IRES-GFP into bone marrow cells of the Trib1 knockout mouse. The Hoxa9-expressing cells with (Trib1 hi) or without (Trib1 null) Trib1 expression showed immature myeloid lineage morphologies (Figure 1A). Both cell types were positive for Mac1 and Gr1, yet Trib1 hi cells showed decreased Mac1 and Gr1 expression levels and increased CD34 expression (Figure 1B), suggesting more immature characteristics of Trib1 hi than Trib1 null cells. Previous studies showed that Trib1 induces COP1-dependent degradation of the C/EBPα p42 isoform and enhancement of ERK phosphorylation by interacting with MEK1.13,14,24 Selective loss of the p42 isoform of C/EBPα is important for leukemogenesis.25-27 In agreement, C/EBPα p42 but not p30 was found significantly decreased and ERK phosphorylation was enhanced and prolonged upon interleukin-3 stimulation in Trib1 hi cells (Figure 1C-D). Trib1 hi cells also showed higher proliferation rates and 5-ethynyl-2′-deoxyuridine incorporation than those of Trib1 null cells (Figure 1E-F), indicating that Trib1 induces increased cell cycling. Indeed, gene microarray analysis of Trib1 hi and null cells showed a distinct expression profile and Trib1 hi–specific enrichment of cell cycle and C/EBPα pathways, consistent with above results (Figure 1G). We further performed a bone marrow transplantation experiment and found that only Trib1 hi cells could induce in vivo leukemogenesis, as evidenced by the interaction between Hoxa9 and Meis1 (Figure 1H)3 ; as previously mentioned, Hoxa9 overexpression is not sufficient for inducing leukemogenesis in vivo.3,28 Thus, these results indicate that Hoxa9-induced leukemogenesis is promoted by Trib1 overexpression.
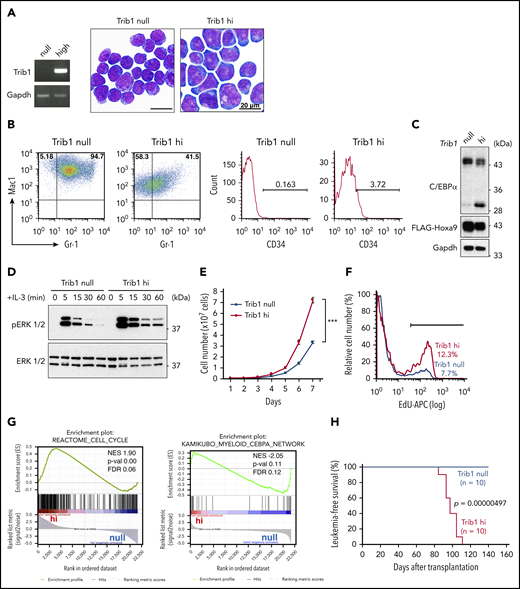
Trib1 expression induces an aggressive AML phenotype. (A) Morphologies of Hoxa9-expressing AML cells with (hi) or without (null) Trib1 expression (right). Reverse transcription (RT) PCR of Trib1 expression in hi cells (left). Scale bars, 20 μm. (B) Fluorescence-activated cell sorting analysis shows decreased expression of Mac1 and Gr-1 (left) and an increased CD34-positive fraction (right) in Trib1 hi cells. The numbers indicate frequencies (%) of Mac1/Gr-1-double-positive, Mac1-single-positive, and CD34-positive fractions. (C) Decreased expression of the p42 isoform of C/EBPα in Trib1 hi cells. (D) Enhanced and prolonged phosphorylation of ERK1/2 in Trib1 hi cells. (E) Increased proliferation of Trib1 hi cells. ***P < .001. (F) Increased 5-ethynyl-2′-deoxyuridine (EdU) incorporation in Trib1 hi cells. (G) Gene set enrichment analysis shows correlation of the cell cycle pathway (left) and inverse correlation of the C/EBPα network gene sets (right). FDR, false discovery rate; NES, normalized enrichment score; p-val, P value. (H) AML developed by transplantation of Trib1 hi cells with a median survival time of 97.1 days, whereas transplantation of Trib1 null cells failed to show AML development in vivo. Significance between 2 cohorts was examined by a log-rank test.
Trib1 expression induces an aggressive AML phenotype. (A) Morphologies of Hoxa9-expressing AML cells with (hi) or without (null) Trib1 expression (right). Reverse transcription (RT) PCR of Trib1 expression in hi cells (left). Scale bars, 20 μm. (B) Fluorescence-activated cell sorting analysis shows decreased expression of Mac1 and Gr-1 (left) and an increased CD34-positive fraction (right) in Trib1 hi cells. The numbers indicate frequencies (%) of Mac1/Gr-1-double-positive, Mac1-single-positive, and CD34-positive fractions. (C) Decreased expression of the p42 isoform of C/EBPα in Trib1 hi cells. (D) Enhanced and prolonged phosphorylation of ERK1/2 in Trib1 hi cells. (E) Increased proliferation of Trib1 hi cells. ***P < .001. (F) Increased 5-ethynyl-2′-deoxyuridine (EdU) incorporation in Trib1 hi cells. (G) Gene set enrichment analysis shows correlation of the cell cycle pathway (left) and inverse correlation of the C/EBPα network gene sets (right). FDR, false discovery rate; NES, normalized enrichment score; p-val, P value. (H) AML developed by transplantation of Trib1 hi cells with a median survival time of 97.1 days, whereas transplantation of Trib1 null cells failed to show AML development in vivo. Significance between 2 cohorts was examined by a log-rank test.
DNA-binding properties of Hoxa9 and C/EBPα in AML
As overlapping DNA binding of Hoxa9 and C/EBPα was reported, we aimed to examine the global binding of Hoxa9 and C/EBPα in the presence or absence of Trib1 expression. Overall distribution of Hoxa9 DNA-binding peaks were not significantly different between Trib1 null and hi cells, as 39% or 27% of DNA-binding peaks were located within 2 kb of transcription start sites (TSSs), respectively (Figure 2A). MEME suite motif analysis for both Hoxa9- (FLAG) and C/EBPα-binding peaks showed a significant concentration of Hoxa9, C/EBPα, and Meis1 (Figure 2B), indicating that Hoxa9 and C/EBPα are close to each another at the chromatin level. Frequent associations in DNA binding between Hoxa9 and C/EBPα were observed in Trib1 null cells, with a 60.5% overlap in Hoxa9-binding peaks with those of C/EBPα (Figure 2C). As Hoxa9 reprograms the enhancer profile in leukemia,29 we compared the distribution of histone H3K27ac and Hoxa9 binding. We found that Hoxa9 and H3K27ac peaks were frequently associated together in both Trib1 null and hi cells (Figure 2D), while the global deposition of H3K27ac was not very different between Trib1 null and hi cells. Codepositions of Hoxa9, C/EBPα, and H3K27ac were detected along the promoter, intragenic, and intergenic regions of Trib1 hi and null cells (Figure 2E). In addition, DNA binding of C/EBPα in Trib1 hi cells in which the p30 isoform is enriched due to degradation of p42 was examined by ChIP-seq. The results indicate a 41.1% overlap of C/EBPα-binding sites between hi and null cells (Figure 2E; supplemental Figure 1A-B), suggesting that p30 may play a different role in hi cells.
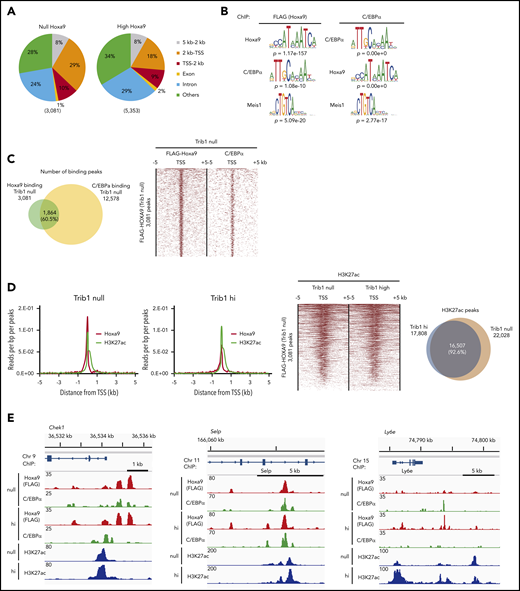
Hoxa9-binding sites in Trib1 hi and null cells show close association with enhancers and C/EBPα-binding sites. (A) Global distribution of Hoxa9-binding peaks. (B) MEME suite motif analysis shows enrichment of the C/EBPα motif in Hoxa9-binding peaks and vice versa in Trib1 null cells. The Meis1-binding motif was also enriched in both Hoxa9 and C/EBPα peaks. (C) Venn diagram showing frequent overlaps between Hoxa9- and C/EBPα-binding peaks in Trib1 null cells (left). Density plot of Hoxa9 and C/EBPα ChIP-seq data sets centered on TSSs (right). Each row represents a single peak. (D) Meta-profile of average ChIP-seq signals for Hoxa9 and H3K27ac in Trib1 null (left) and hi (right) cells in the region ± 5 kb around TSSs. (E) ChIP-seq occupancy profiles for C/EBPα, Hoxa9, and H3K27ac at the Chek1, Selp, and Ly6e loci.
Hoxa9-binding sites in Trib1 hi and null cells show close association with enhancers and C/EBPα-binding sites. (A) Global distribution of Hoxa9-binding peaks. (B) MEME suite motif analysis shows enrichment of the C/EBPα motif in Hoxa9-binding peaks and vice versa in Trib1 null cells. The Meis1-binding motif was also enriched in both Hoxa9 and C/EBPα peaks. (C) Venn diagram showing frequent overlaps between Hoxa9- and C/EBPα-binding peaks in Trib1 null cells (left). Density plot of Hoxa9 and C/EBPα ChIP-seq data sets centered on TSSs (right). Each row represents a single peak. (D) Meta-profile of average ChIP-seq signals for Hoxa9 and H3K27ac in Trib1 null (left) and hi (right) cells in the region ± 5 kb around TSSs. (E) ChIP-seq occupancy profiles for C/EBPα, Hoxa9, and H3K27ac at the Chek1, Selp, and Ly6e loci.
Trib1 modulates super-enhancer profiles in Hoxa9-induced AML
Super-enhancers control cell-lineage–specific genes, define cell identities,30 and regulate important oncogenic pathway genes.21,31 Because Hoxa9 was closely associated with enhancer marks, super-enhancer profiles were compared between Trib1 hi and null cells to examine whether Trib1 can modify super-enhancers. After analyzing normalized H3K27Ac signals, 437 and 765 super-enhancers were identified that were associated with 63.1% and 46.2% of Hoxa9 binding loci in Trib1 hi and null cells, respectively (Figure 3A; supplemental Table 4). Systemic analysis of biological process pathways for Trib1 hi- or null-specific super-enhancers identified immune system, hematopoiesis and myeloid cell differentiation pathways in Trib1 hi cells, suggesting important roles for Trib1 hi–specific super-enhancers in the hematopoietic system (Figure 3B; supplemental Figure 2). To identify important targets of the modified super-enhancer activity regulated by Trib1 and Hoxa9, we selected 61 Trib1 hi–specific super-enhancers. Among these, 30 were associated with Hoxa9 binding (supplemental Table 4), and 8 genes were found to be upregulated (fold change > 2.0) in Trib1 hi cells and identified as significantly modified genes by Trib1 overexpression (Figure 3C). The significant increase in mRNA expression levels of these 8 genes (Erg, Spns2, Rgl1, Tbc1d9b, Pik3cd, Lfng, Atg7, and Hspa4) was validated by quantitative RT-PCR (Figure 3D). ChIP-quantitative PCR for these loci showed a significant increase in H3K27Ac depositions at the Erg, Spns2, Rgl1, and Pik3cd loci in Trib1 hi cells (Figure 3E; supplemental Figures 3A and 4). Among these genes, a remarkable difference of H3K27Ac between Trib1 hi and null cells was observed at the Erg locus including its +85-stem cell enhancer (Figure 3F).32 CRISPR/Cas9-mediated targeting of the Erg +85 enhancer was carried out. From this, a homozygous deletion of 50 bp, including a putative Hoxa9-binding site within the enhancer, abolished upregulation of Erg (Figure 3G; supplemental Figure 5). This strongly suggests that Erg is upregulated by Hoxa9 binding at the +85 enhancer.
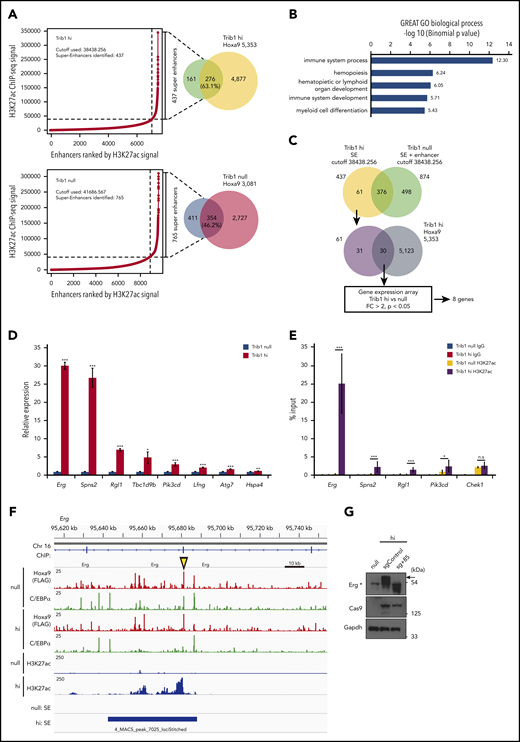
Different super-enhancer distribution between Trib1 hi and null cells identifies Hoxa9/Trib1 target genes. (A) Enhancers were ranked by increasing H3K27ac ChIP-seq signals in Trib1 hi (left, top) and null (left, bottom) cells. Using the ROSE algorithm, 437 and 765 enhancers were defined as super-enhancers in Trib1 hi and null cells, respectively. Overlap between super-enhancers and Hoxa9 DNA-binding peaks are shown in Venn diagrams (right). (B) Enrichment of Gene Ontology biological process for Trib1 hi super-enhancer loci. (C) Schematic diagram for target gene identification. SE, super-enhancer. (D) Quantitative RT-PCR shows increased expression of Hoxa9/Trib1 target genes in Trib1 hi cells. (E) Quantitative ChIP-PCR of H3K27ac signals for super-enhancers of Erg, Spns2, Rgl1, and Pik3cd. The Chek1 locus is shown as a negative control. Three distinct loci for each super-enhancer were examined, and the average accumulation in these 3 loci is indicated. (F) Density plots for ChIP-seq reads of C/EBPα, Hoxa9, and H3K27ac in Trib1 hi and null cells at the +85 enhancer of Erg. The yellow arrowhead indicates the position of the sgRNA for the +85 enhancer. (G) Immunoblotting shows significant decrease of Erg protein expression (arrow) by knockout of the Erg enhancer using a sgRNA for the +85 enhancer. The asterisk in immunoblotting indicates nonspecific bands. *P < .05, **P < .01, ***P < .001; n.s, not significant.
Different super-enhancer distribution between Trib1 hi and null cells identifies Hoxa9/Trib1 target genes. (A) Enhancers were ranked by increasing H3K27ac ChIP-seq signals in Trib1 hi (left, top) and null (left, bottom) cells. Using the ROSE algorithm, 437 and 765 enhancers were defined as super-enhancers in Trib1 hi and null cells, respectively. Overlap between super-enhancers and Hoxa9 DNA-binding peaks are shown in Venn diagrams (right). (B) Enrichment of Gene Ontology biological process for Trib1 hi super-enhancer loci. (C) Schematic diagram for target gene identification. SE, super-enhancer. (D) Quantitative RT-PCR shows increased expression of Hoxa9/Trib1 target genes in Trib1 hi cells. (E) Quantitative ChIP-PCR of H3K27ac signals for super-enhancers of Erg, Spns2, Rgl1, and Pik3cd. The Chek1 locus is shown as a negative control. Three distinct loci for each super-enhancer were examined, and the average accumulation in these 3 loci is indicated. (F) Density plots for ChIP-seq reads of C/EBPα, Hoxa9, and H3K27ac in Trib1 hi and null cells at the +85 enhancer of Erg. The yellow arrowhead indicates the position of the sgRNA for the +85 enhancer. (G) Immunoblotting shows significant decrease of Erg protein expression (arrow) by knockout of the Erg enhancer using a sgRNA for the +85 enhancer. The asterisk in immunoblotting indicates nonspecific bands. *P < .05, **P < .01, ***P < .001; n.s, not significant.
C/EBPα degradation by Trib1 is important for enhancer reprograming and Hoxa9 target gene expression
Previous studies indicated that Trib1 promotes C/EBPα p42 degradation and enhances ERK phosphorylation using different binding motifs to Cop1 and MEK1.14 To determine which pathway is important for enhancer reprograming and Hoxa9 target gene expression, we inhibited MEK1 in Trib1 hi cells or knocked down Cebpa in Trib1 null cells, as Trib1 hi cells could not survive by knockdown of MEK1. Suppression of H3K27Ac accumulation in super-enhancers at the Erg, Spns2, and Pik3cd loci was observed in Trib1 hi cells treated with the MEK1 inhibitor U0126 (Figure 4A; supplemental Figure 3B); however, downregulation of mRNA expression was only marginal (Figure 4B). In contrast, H3K27Ac signals at the Erg, Spns2, Rgl1, and Pik3cd super-enhancers were significantly increased after Cebpa silencing in Trib1 null cells (Figure 4C; supplemental Figure 3C). Moreover, expression of these 4 genes was remarkably upregulated (Figure 4D). Next, the isoform-specific C/EBPα function was tested by introducing human CEBPA cDNAs encoding full-length (p42 and p30) or N-terminus-truncated (p30 only) proteins into Cebpa-silenced Trib1 null cells to avoid shRNA-mediate gene silencing. The results showed the expression of Erg mRNA and protein as well as accumulation of the histone H3K27Ac were partially recovered by p42, but not p30, expression (Figure 4E; supplemental Figure 3D). Together, these results indicate that C/EBPα p42 degradation by Trib1 is a major driving force for enhancer modification at certain Hoxa9-binding loci and that MEK/ERK enhancement contributes slightly to this event.
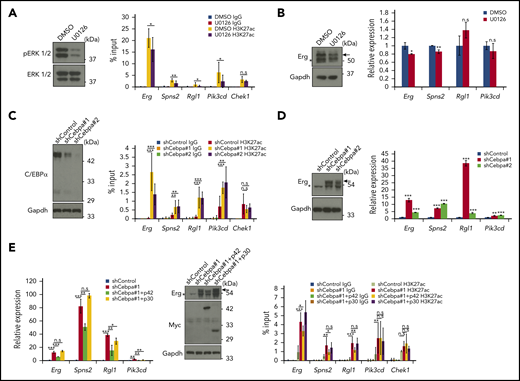
Trib1 modulates super-enhancer activity and gene expression via C/EBPα degradation. (A) Trib1 hi cells were treated with the MEK1 inhibitor U0126 (10 μM) for 24 hours. Inhibition of ERK1/2 phosphorylation is evidenced by immunoblotting (left). Quantitative ChIP-PCR showing relative signals of H3K27ac to input DNA (right). The Chek1 locus was used as a negative control. (B) Quantitative RT-PCR showing mild downregulation of Erg and Spns2 expression by U0126 treatment (right). Expression of the Erg protein (arrow) was also diminished (top). (C) Cebpa was silenced by shRNA treatment using 2 different sequences in Trib1 null cells. The effect of shRNA was confirmed by immunoblotting (left). Quantitative ChIP-PCR showing relative signals of H3K27ac to input DNA (right). The Chek1 locus was used as a negative control. Three distinct loci for each super-enhancer were examined and the average accumulation in these 3 loci is indicated. (D) Quantitative RT-PCR showing upregulation of Erg, Spns2, Rgl1, and Pik3cd expression by Cebpa silencing (right). Expression of the Erg protein (arrow) is significantly upregulated (left). (E) Quantitative RT-PCR showing partial reduction of the Cebpa silencing effect on Erg, Spns2, Rgl1, and Pik3cd expression by human CEBPA p42, but not p30, expression (left). Western blot showing moderate decrease of the Erg protein by p42, but not p30, expression (center, top). Myc-tagged p42 or p30 expression (center, middle). Quantitative ChIP-PCR showing relative signals of H3K27ac to input DNA (right). The Chek1 locus was used as a negative control. *P < .05, **P < .01, ***P < .001. DMSO, dimethyl sulfoxide.
Trib1 modulates super-enhancer activity and gene expression via C/EBPα degradation. (A) Trib1 hi cells were treated with the MEK1 inhibitor U0126 (10 μM) for 24 hours. Inhibition of ERK1/2 phosphorylation is evidenced by immunoblotting (left). Quantitative ChIP-PCR showing relative signals of H3K27ac to input DNA (right). The Chek1 locus was used as a negative control. (B) Quantitative RT-PCR showing mild downregulation of Erg and Spns2 expression by U0126 treatment (right). Expression of the Erg protein (arrow) was also diminished (top). (C) Cebpa was silenced by shRNA treatment using 2 different sequences in Trib1 null cells. The effect of shRNA was confirmed by immunoblotting (left). Quantitative ChIP-PCR showing relative signals of H3K27ac to input DNA (right). The Chek1 locus was used as a negative control. Three distinct loci for each super-enhancer were examined and the average accumulation in these 3 loci is indicated. (D) Quantitative RT-PCR showing upregulation of Erg, Spns2, Rgl1, and Pik3cd expression by Cebpa silencing (right). Expression of the Erg protein (arrow) is significantly upregulated (left). (E) Quantitative RT-PCR showing partial reduction of the Cebpa silencing effect on Erg, Spns2, Rgl1, and Pik3cd expression by human CEBPA p42, but not p30, expression (left). Western blot showing moderate decrease of the Erg protein by p42, but not p30, expression (center, top). Myc-tagged p42 or p30 expression (center, middle). Quantitative ChIP-PCR showing relative signals of H3K27ac to input DNA (right). The Chek1 locus was used as a negative control. *P < .05, **P < .01, ***P < .001. DMSO, dimethyl sulfoxide.
Erg is an important downstream target involved in the effect of Trib1 on Hoxa9
ChIP-seq and expression analyses showed upregulation of Erg by Trib1 overexpression on the mRNA level. In accordance with these results, expression of the Erg protein p55 isoform that is required for target gene regulation of Erg33 was also upregulated in Trib1 hi cells (Figure 5A). Erg encodes an ETS family transcription factor that is essential for hematopoiesis and vasculogenesis,34,35 and is also involved in human AML.36-38 To clarify the functional role of Erg in Hoxa9-induced AML, Erg was expressed in Trib1 null cells. Trib1 null cells expressing Erg showed increased cellular proliferation comparable to that of Trib1 hi cells (Figure 5B). Conversely, shRNA-mediated Erg knockdown in Trib1 hi cells showed growth suppression (Figure 5C), and CRISPR/Cas9-mediated silencing of Erg further showed growth inhibition of Trib1 hi cells equivalent to that of null cells (Figure 5D). Finally, Erg deletion completely abolished leukemia development of Trib1 hi cells in vivo (Figure 5E), though Erg overexpression in Trib1 null cells did not develop leukemia, indicating that Erg upregulation is required, but not sufficient, for in vivo leukemogenesis in Trib1-enhanced AML associated with Hoxa9 expression. The absence of FLAG-Hoxa9–positive fractions in recipients transplanted with sgErg-silenced Trib1 hi cells suggests decreased activity of engraftments (supplemental Figure 6). However, the exact promotion mechanism of in vivo leukemogenesis by Erg remains to be clarified.
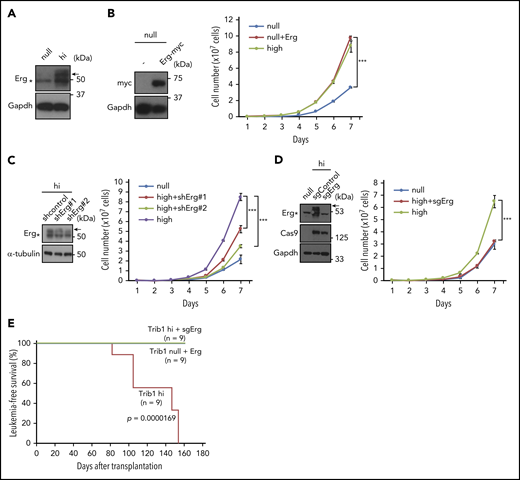
Erg is an important downstream target involved in the effect Trib1 has on Hoxa9. (A) Immunoblotting showing an increase in 55-kDa Erg protein expression (arrow) in Trib1 hi cells. (B) Growth promotion of Trib1 null cells by Myc-tagged Erg overexpression. Immunoblotting of Myc-tagged Erg protein expression (left). Cell numbers of Trib1 null, Trib1 null with Erg, and Trib1 hi cells are shown on the right. (C) shRNA-mediated silencing of Erg. Relative Erg mRNA expression is shown (left). Cell numbers of Trib1 null, Trib1 hi, and Trib1 hi introduced with 2 different shRNA lentivirus are shown on the right. (D) Erg knockout by CRISPR/Cas9 in Trib1 hi cells. Cell numbers of Trib1 null, Trib1 hi, and Trib1 hi cells with sgRNA-mediated Erg knockout (right). (E) A Kaplan-Meier survival curve for Trib1 hi, Trib1 hi cells with sgRNA-mediated Erg knockout, and Trib1 null cells with Erg overexpression. Significance between Trib1 hi and Trib1/Erg knockout, and between Trib1 hi and Trib1 null/Erg overexpression was examined by a log-rank test. ***P < .001.
Erg is an important downstream target involved in the effect Trib1 has on Hoxa9. (A) Immunoblotting showing an increase in 55-kDa Erg protein expression (arrow) in Trib1 hi cells. (B) Growth promotion of Trib1 null cells by Myc-tagged Erg overexpression. Immunoblotting of Myc-tagged Erg protein expression (left). Cell numbers of Trib1 null, Trib1 null with Erg, and Trib1 hi cells are shown on the right. (C) shRNA-mediated silencing of Erg. Relative Erg mRNA expression is shown (left). Cell numbers of Trib1 null, Trib1 hi, and Trib1 hi introduced with 2 different shRNA lentivirus are shown on the right. (D) Erg knockout by CRISPR/Cas9 in Trib1 hi cells. Cell numbers of Trib1 null, Trib1 hi, and Trib1 hi cells with sgRNA-mediated Erg knockout (right). (E) A Kaplan-Meier survival curve for Trib1 hi, Trib1 hi cells with sgRNA-mediated Erg knockout, and Trib1 null cells with Erg overexpression. Significance between Trib1 hi and Trib1/Erg knockout, and between Trib1 hi and Trib1 null/Erg overexpression was examined by a log-rank test. ***P < .001.
BRD4 inhibitor suppresses Hoxa9-induced leukemia growth in a Trib1-dependent manner
The BET domain containing transcriptional coactivator BRD4 was found abundantly accumulated in super-enhancers.39 In an attempt to target Trib1/Hoxa9–driven super-enhancers, Trib1 hi cells were treated with the BRD4 inhibitor JQ1.21 JQ1 administration efficiently suppressed leukemia cell growth in a Trib1-dependent manner (Figure 6A; supplemental Figure 7) and suppressed expression of the Trib1/Hoxa9 targets Erg and Spns2, but not Rgl1 and Pik3cd, in Trib1 hi cells (Figure 6B). JQ1 treatment also induced granulo-monocytic differentiation and early apoptosis of Trib1 hi cells (Figures 6C-D). The effects of the CDK7/8 inhibitor THZ1 was also examined as a super-enhancer–targeting drug,31 and in contrast to JQ1, THZ1 treatment showed only mild antiproliferative effects, while Erg and Spns2 downregulation was not observed (Figures 6E-F). Finally, we assessed the in vivo antileukemic activity of JQ1 against Trib1 hi cells. Mice were administered JQ1 1 week after bone marrow transplantation of Trib1 hi cells with or without daunorubicin and cytarabine treatment. JQ1 treatment significantly improved the survival of leukemic mice regardless of daunorubicin and cytarabine treatment (Figure 6G). Collectively, the results highlight the importance of targeting the Erg super-enhancer modified by Trib1.
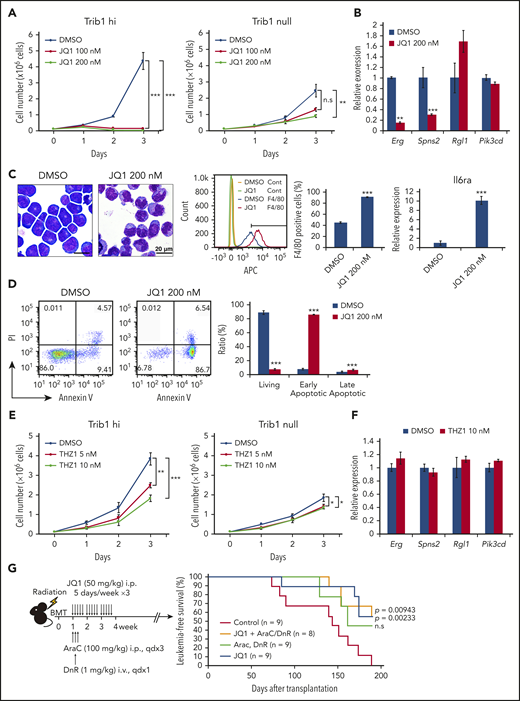
JQ1 treatment inhibits the growth of Trib1 hi AML and leads to Erg downregulation. (A) Growth of Trib1 hi (left) and null (right) cells treated with JQ1 or vehicle. (B) Quantitative RT-PCR showing downregulation of Erg and Spns2 but not Rgl1 and Pik3cd expression after JQ1 treatment. (C) Monocytic differentiation of Trib1 hi cells by JQ1 treatment of 24 hours. Left, Giemsa staining; center, flow cytometric analysis for F4/80 expression. The isotype control-stained cells are indicated as yellow and green histograms. F4/80-positive fractions are quantitated as bar graphs; right, qRT-PCR for Il6ra expression. Scale bar: 20 μm. (D) Detection of early apoptosis induced by JQ1 treatment of 48 hours. Annexin V staining shows a significant increase in early apoptotic cells, as evidenced by flow cytometry (left). Bar graphs show an increase in both early and late apoptotic cells (right). (E) Growth of Trib1 hi (left) and null (right) cells treated with THZ1 or vehicle. (F) Quantitative RT-PCR shows no significant changes in target gene expression by THZ1 treatment. (G) In vivo treatment of mice bearing AML with JQ1 and with or without cytarabine (AraC) and daunorubicin (DnR). Schematic illustration of the experiment (left). Kaplan-Meier survival curves showing improvement of survival with JQ1 treatment and with or without AraC and DnR. ** P < .01, *** P < .001. BMT, bone marrow transplant; i.p., intraperitoneal; PI, propidium iodide.
JQ1 treatment inhibits the growth of Trib1 hi AML and leads to Erg downregulation. (A) Growth of Trib1 hi (left) and null (right) cells treated with JQ1 or vehicle. (B) Quantitative RT-PCR showing downregulation of Erg and Spns2 but not Rgl1 and Pik3cd expression after JQ1 treatment. (C) Monocytic differentiation of Trib1 hi cells by JQ1 treatment of 24 hours. Left, Giemsa staining; center, flow cytometric analysis for F4/80 expression. The isotype control-stained cells are indicated as yellow and green histograms. F4/80-positive fractions are quantitated as bar graphs; right, qRT-PCR for Il6ra expression. Scale bar: 20 μm. (D) Detection of early apoptosis induced by JQ1 treatment of 48 hours. Annexin V staining shows a significant increase in early apoptotic cells, as evidenced by flow cytometry (left). Bar graphs show an increase in both early and late apoptotic cells (right). (E) Growth of Trib1 hi (left) and null (right) cells treated with THZ1 or vehicle. (F) Quantitative RT-PCR shows no significant changes in target gene expression by THZ1 treatment. (G) In vivo treatment of mice bearing AML with JQ1 and with or without cytarabine (AraC) and daunorubicin (DnR). Schematic illustration of the experiment (left). Kaplan-Meier survival curves showing improvement of survival with JQ1 treatment and with or without AraC and DnR. ** P < .01, *** P < .001. BMT, bone marrow transplant; i.p., intraperitoneal; PI, propidium iodide.
Involvement of the TRIB1/ERG axis in human AML cells
To evaluate the significance of the TRIB1/ERG axis as well as possible super-enhancer involvement, TRIB1 and HOXA9 expression was analyzed in human AML cell lines (Figure 7A; supplemental Table 5). Among the cell lines, KU812 and P39 showed high levels of TRIB1 and HOXA9 expression and were subjected to further examination. Low levels of ERG expression in HL-60 cells suggests that these cells may possess different regulatory mechanisms of ERG expression by HOXA9. shRNA-mediated knockdown of TRIB1 showed downregulation of ERG and SPNS2 in KU812 cells (Figure 7B). Moreover, growth suppression was observed in KU812 cells after shRNA-mediated TRIB1 knockdown (Figure 7C). Next, JQ1 treatment at 250 nM showed growth suppression effects on KU812 and P39 cells (Figure 7D) and suppressed ERG expression in both cell lines (Figure 7E). These findings indicate that TRIB1-mediated ERG upregulation plays an important role in leukemic cell growth in human AML and that BRD4 inhibition by JQ1 is a potential therapeutic strategy. Furthermore, it has been reported that overexpression of ERG predicts worse outcomes in of normal-karyotype AML.40 Thus, we reassessed the possible roles of ERG and SPNS2 upregulation in human AML by the PrognoScan online platform (http://www.prognoscan.org/)41 and found that high expression levels of ERG or SPNS2 were significantly correlated with poor prognosis in normal-karyotype AML (Figure 7F). Correlative expression between HOXA9 and TRIB1, but not TRIB2, was evident in the same cohort, although there was no correlation between ERG and TRIB or TRIB2. This was probably due to the accumulation of multiple genetic mutations in human cases (supplemental Figure 8). Collectively, our results indicate that ERG is an important target of Hoxa9 and C/EBPα and is modulated by Trib1. The role of SPNS2 in myeloid leukemogenesis remains to be clarified, although it has a role in STAT3-induced carcinogenesis by promoting inflammatory conditions in the tumor microenvironment.42
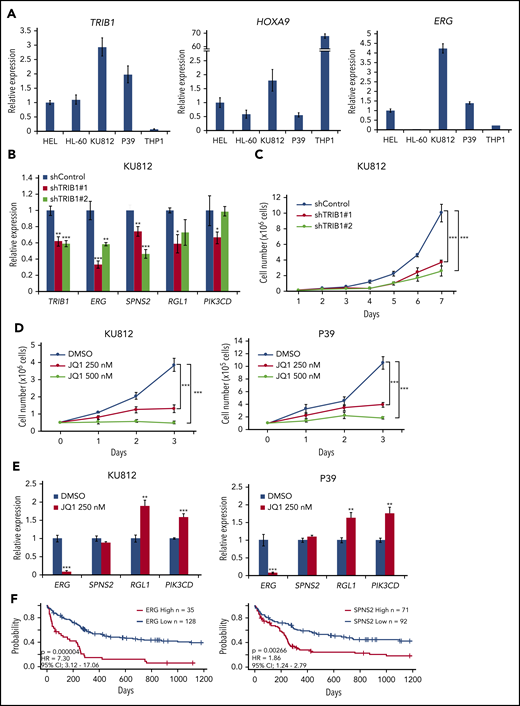
An important role for the TRIB1/ERG axis in human AML. (A) Quantitative RT-PCR for TRIB1, HOXA9, and ERG in human AML cell lines. (B) Downregulation of TRIB1/HOXA9 downstream genes in KU812 cells by TRIB1 silencing. Relative gene expression to shControl in KU812 cells. (C) Growth inhibition by TRIB1 knockdown. Cell numbers of KU812 cells treated with 2 different shRNA lentivirus. (D) KU812 and P39 cells were treated with JQ1 at the indicated dosages for 3 days. Cell numbers at the indicated time points are shown. (E) Effects of JQ1 on gene expression in KU812 and P39 cells. Relative mRNA expression of the indicated genes is shown. (F) Kaplan-Meier survival curves for patients for patients with normal-karyotype AML64 between high and low expression of ERG (left) or SPNS2 (right). Significances were measured by a log-rank test. Hazard ratios (HRs) and 95% confidence intervals (CIs) are indicated. *P < .05, **P < .01, ***P < .001.
An important role for the TRIB1/ERG axis in human AML. (A) Quantitative RT-PCR for TRIB1, HOXA9, and ERG in human AML cell lines. (B) Downregulation of TRIB1/HOXA9 downstream genes in KU812 cells by TRIB1 silencing. Relative gene expression to shControl in KU812 cells. (C) Growth inhibition by TRIB1 knockdown. Cell numbers of KU812 cells treated with 2 different shRNA lentivirus. (D) KU812 and P39 cells were treated with JQ1 at the indicated dosages for 3 days. Cell numbers at the indicated time points are shown. (E) Effects of JQ1 on gene expression in KU812 and P39 cells. Relative mRNA expression of the indicated genes is shown. (F) Kaplan-Meier survival curves for patients for patients with normal-karyotype AML64 between high and low expression of ERG (left) or SPNS2 (right). Significances were measured by a log-rank test. Hazard ratios (HRs) and 95% confidence intervals (CIs) are indicated. *P < .05, **P < .01, ***P < .001.
Discussion
Trib1 encodes a pseudokinase that lacks kinase activities and instead acts as a molecular adaptor in critical signaling pathways, such as the ubiquitination of C/EBP transcription factors and CDC25, and interacts with MEKs to modulate downstream signals.10,43 In the myeloid lineage, Trib1 exhibits a strong oncogenic activity by degrading C/EBPα p42 and enhancing ERK phosphorylation.10,13,14 In addition, overexpression of Trib1 significantly accelerates Hoxa9-induced leukemogenesis,1 suggesting that Trib1 reinforces the transcriptional activity of Hoxa9. By analyzing the global DNA binding of Hoxa9 and C/EBPα as well as histone H3K27Ac levels, we clarified the modulation of Hoxa9-associated super-enhancers via Trib1 overexpression. Silencing of Cebpa showed significant reduction of H3K27Ac signals at these super-enhancers, whereas MEK inhibition only slightly contributed to the reduction, indicating that removal of C/EBPα from the super-enhancers is an important function of Trib1 in enhancer modification.
C/EBPα is a myeloid tumor suppressor25-27 ; however, null mutations of CEBPA do not induce AML.44 Selective loss of the p42 isoform of C/EBPα is important for Trib2-induced leukemogenesis,45,46 and Trib1 interacts exclusively with the p42, but not p30, isoform.47 In agreement with this, p30 expression was preserved in Trib1 hi cells (Figure 1C) or even increased due to a negative-feedback mechanism, as evidenced by microarray analysis. Increased Hoxa9 binding at the Cebpa enhancer suggests that Hoxa9 may play some role in the enhancement of Cebpa transcription (supplemental Figure 9). Our ChIP-seq results on the Trib1 hi cells indicated that 41.1% of the C/EBPα binding sites were preserved, suggesting that p30 could, in part, replace DNA binding of p42. However, reduced binding at the Erg enhancer and loss of the N-terminal transactivation domain might cause reduced histone K27 acetylation and target gene expression. C/EBPα is also required for the initiation of MLL fusion–mediated and Hoxa9/Meis1–expressing AML development,48 suggesting that C/EBPα recruits Hoxa9 and/or Meis1 to their target sites.49 Indeed, the present study and others14,15 showed frequent overlaps in DNA binding between C/EBPα and Hoxa9. It is reported that the p30, but not p42, isoform interacts with a trithorax component WDR5,50 and this interaction could induce trithorax-dependent H3K27 acetylation, though further studies are needed to clarify the underlying mechanisms. In addition to C/EBPα degradation, Trib1 affects the MEK/ERK pathway by binding MEK1,14 as was confirmed by enhanced ERK phosphorylation in Trib1 hi cells (Figure 1D). However, inhibition of this pathway only mildly affects super-enhancer modification. Nevertheless, C/EBPα phosphorylation induced by phosphorylated ERK diminishes homodimerization and DNA binding of C/EBPα,51 suggesting that increased activation of the MEK/ERK pathway by Trib1 may also contribute to C/EBPα downregulation.
Erg is an ETS family transcription factor that is critical for hematopoiesis, angiogenesis, and chondrogenesis during embryogenesis and gestation.34,35,52 Upregulation of ERG in human AML patients was reported, and its important role in leukemic cell maintenance was evident.53,54 Previous studies reported that ERG is a downstream target of HOXA9 and that overexpression of ERG is an important biomarker in predicting poor prognoses for patients with normal-karyotype AML16,40 ; this is in agreement with our findings. Other studies showed that a heptad of transcription factors (FLI1, ERG, GATA2, RUNX1, SCL/TAL1, LYL1, and LMO2) recognize the ERG enhancer 85 kb downstream of a translation initiation site (+85 enhancer)32,55,56 that is well conserved between humans and mice and is a target of Trib1 modification, as revealed by our study. Core transcription factor binding on the +85 enhancer indicates the presence of a core regulatory circuitry (CRC) that is important for the super-enhancer activity as well as hematopoietic differentiation and transformation.57,58 Modification of the +85 Erg super-enhancer by Trib1 overexpression suggests that Trib1 promotes the activities of CRCs and super-enhancers, resulting in Erg upregulation and more malignant AML phenotypes. Upregulation of Erg was also induced by Trib2 overexpression in Trib1 null cells (supplemental Figure 10), suggesting that Trib2 could promote the +85 super-enhancer activity. Although Erg is not the only target gene that Hoxa9 and Trib1 share, having Spns2, Rgl1, and Pik3cd as other targets. Erg is the most significantly downregulated gene by Trib1 silencing and knockout/knockdown of Erg is critical for the growth of Trib1 hi cells. These results indicate the critical role of Erg in the aggressive phenotype accompanied by upregulated Trib1.
The increased sensitivity of Trib1 hi cells to the BRD4 inhibitor JQ1 is consistent with the close association of BRD4 chromatin occupancy with CRC-associated transcription factors such as ERG and C/EBPα.39 JQ1 treatment suppresses the super-enhancer activity, including the Erg +85 locus, to suppress transcription of Erg mRNA. Moreover, it also inhibits the ERG and BRD4 association to abrogate transcriptional activation of ERG target genes.39 Thus, Trib1 overexpression may be a potential biomarker in predicting improved responses to BRD4 inhibitors. JQ1 treatment showed a mild suppressive growth effect on Trib1 null cells probably by targeting common super-enhancers present both in Trib1 hi and null cells. However, the apparent discrepancy of SPNS2, RGL1, and PIK3CD responses to JQ1 between mouse and human AML cells remains to be clarified, as differences in the mutational background may affect the expression of these genes. In contrast, the CDK7/8 inhibitor THZ1 failed to show Trib1-specific growth inhibitory effects, indicating that the CDK7/8/c-MYC axis is not directly linked to the Hoxa9-regulated transcriptional program in the maintenance of AML.59 There may be other fine-tuning mechanisms of Hoxa9-driven super-enhancer activity, and the presence of C/EBPα may alter the activity of the certain loci; thus, further research is warranted.
Altogether, our findings clarified the significance and molecular mechanisms underlying the effect Trib1 has on Hoxa9 interaction in myeloid leukemogenesis. The importance of tribbles family pseudokinases as therapeutic targets has been emphasized not only in leukemia but also in other malignancies, including breast, prostate, and colorectal cancers.60-62 Recent studies targeting Trib1 via small-molecule inhibitors or Trib1 by conventional kinase inhibitors highlighted targeting tribbles psuedokinases as an attractive method for developing novel therapeutic strategies against cancers that are resistant to conventional treatment.24,63
The microarray and ChIP-seq data reported in this article have been deposited in the NCBI Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo) (accession numbers GSE139641 and GSE140313, respectively).
For original data, please contact takuro-ind@umin.net.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Hironori Harada for human AML cell lines and Toshio Kitamura for human CEBPA cDNAs.
This work was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (19H01035 to T.N. and 18K15227 to S.Y.) and by a Grant-in-Aid for Cyclic Innovation for Clinical Empowerment from Japan Agency for Medical Research and Development (A0101033) (T.N.).
Authorship
Contribution: S.Y., T.Y., Y.S., T.T., A.N., and Y.Y. performed the research and interpreted and analyzed the data; S.T. and H.A. performed genomic and bioinformatic analyses; and T.N designed and supervised the study and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Takuro Nakamura, Division of Carcinogenesis, The Cancer Institute, Japanese Foundation for Cancer Research, 3-8-31 Ariake, Koto-ku, Tokyo 135-8550, Japan; e-mail: takuro-ind@umin.net.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal