

Key Points
Donor-derived T cells with native specificity for multiple myeloid leukemia antigens can be expanded ex vivo.
Infusion of mLSTs after HCT is well tolerated and produces antileukemia effects.

Abstract
Relapse after allogeneic hematopoietic stem cell transplantation (HCT) is the leading cause of death in patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS). Infusion of unselected donor lymphocytes (DLIs) enhances the graft-versus-leukemia (GVL) effect. However, because the infused lymphocytes are not selected for leukemia specificity, the GVL effect is often accompanied by life-threatening graft-versus-host disease (GVHD), related to the concurrent transfer of alloreactive lymphocytes. Thus, to minimize GVHD and maximize GVL, we selectively activated and expanded stem cell donor–derived T cells reactive to multiple antigens expressed by AML/MDS cells (PRAME, WT1, Survivin, and NY-ESO-1). Products that demonstrated leukemia antigen specificity were generated from 29 HCT donors. In contrast to DLIs, leukemia-specific T cells (mLSTs) selectively recognized and killed leukemia antigen–pulsed cells, with no activity against recipient's normal cells in vitro. We administered escalating doses of mLSTs (0.5 to 10 × 107 cells per square meter) to 25 trial enrollees, 17 with high risk of relapse and 8 with relapsed disease. Infusions were well tolerated with no grade >2 acute or extensive chronic GVHD seen. We observed antileukemia effects in vivo that translated into not-yet-reached median leukemia-free and overall survival at 1.9 years of follow-up and objective responses in the active disease cohort (1 complete response and 1 partial response). In summary, mLSTs are safe and promising for the prevention and treatment of AML/MDS after HCT. This trial is registered at www.clinicaltrials.com as #NCT02494167.
Introduction
Posttransplantation relapse of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) is a devastating diagnosis with very few effective treatment options. Cellular therapy in the form of donor lymphocyte infusions (DLIs) and/or a second allogeneic hematopoietic stem cell transplantation (HCT) have been more effective than chemotherapy alone in treating post-HCT relapse.1-4 However, the toxicities of conditioning chemotherapy and graft-versus-host disease (GVHD) and the need for chronic immunosuppression are significant and life threatening, greatly limiting the number of eligible patients. Furthermore, only a minority of relapsed patients (<30%) will achieve long-term remission after DLI or second HCT.3,5 Thus, there is a clear need to minimize toxicities and to improve the antileukemia effects of available cellular immunotherapies after HCT.
To develop a safe and effective cellular therapy for patients with relapsed AML/MDS, we designed a T-cell therapy that targets multiple leukemia-associated antigens and thereby attempts to mimic the graft-versus-leukemia (GVL) effect mediated by donor T cells, but with a lower risk of inducing GVHD. To this end, we took peripheral blood from hematopoietic stem cell (HSC) donors and expanded naturally occurring T cells whose native T-cell receptors (TCRs) were specific for leukemia-expressed antigens (WT1, PRAME, NY-ESO-1, and Survivin). Herein, we report on the safety and the clinical effects mediated by these stem cell donor–derived multiple leukemia antigen–specific T cells (mLSTs) when infused into 25 HCT recipients with AML/MDS, 17 of whom were at high risk of relapse (in complete response [CR] at the time of infusion, including 5 who had relapsed after HCT but were back in CR after salvage therapy immediately before infusion) and 8 of whom had active AML in which HCT and subsequent salvage treatments had failed.
Methods
Patients
Patients with AML or MDS after HCT were eligible for a Baylor College of Medicine Institutional Review Board–approved protocol (H-36346) to treat refractory/relapsed disease (active arm) or to maintain remission after HCT (adjuvant arm). Inclusion criteria were (1) life expectancy of ≥6 weeks, (2) HCT performed at our center, (3) Karnofsky (or Lansky) score ≥50, (4) provision of informed consent, (5) adequate organ function (bilirubin, serum creatinine ≤2 times and aspartate aminotransferase ≤3 times the upper limit of normal, hemoglobin ≥7 g/dL, and pulse oximetry >90% on room air), (6) use of birth control, (7) available mLSTs, and (8) cessation of investigational antineoplastic therapy for ≥1 month. Exclusion criteria were (1) grade ≥2 acute or extensive stage chronic GVHD, (2) use of corticosteroids ≥0.5 mg/kg prednisone equivalent, (3) use of anti-T-cell antibodies (eg, alemtuzumab) within ≤30 days of infusion, (4) ongoing infection, and (5) pregnancy. Calcineurin inhibitors such as tacrolimus were permitted. Patients had to have attained engraftment, and time since HCT had to be at least 30 days. None of the enrolled patients received lymphodepleting chemotherapy before receiving mLSTs. All patients had to undergo a disease assessment (at a minimum bone marrow examination) within 4 weeks of T-cell infusion. Those with active disease had to have received no therapy for AML/MDS from the time of the bone marrow examination that confirmed relapse or, if they had received intervening therapy, they had to demonstrate treatment failure on infusion day. Bone marrow assessments were repeated at least once after infusion. Once enrolled, patients received a single infusion of mLSTs at one of the dose levels (DLs: DL1, 0.5 × 107, DL2, 1 × 107; DL3, 2 × 107; DL4, 5 × 107, and DL5, 10 × 107 cells per square meter) and were eligible to receive up to 6 additional infusions at the same dose if they remained in complete remission (high-risk arm) or achieved clinical benefit (active arm) at their subsequent evaluation. The dose escalation schema (based on square meters rather than kilograms) closely mirrored doses of Epstein Barr virus–specific T cells that had been shown to be safe and effective in prior clinical trials at our center.6-9 The full protocol is provided in the supplemental Materials, available on the Blood Web site. To compare the frequency of immune evasion tactics at the time of relapse in the mLST-recipient cohort with those who were not treated with mLSTs, a cohort of consecutive adult patients with AML/MDS who had attained successful engraftment by day +30 after matched sibling donor-HCT or DLI at our center from 2012 through 2018 and had not been treated with mLSTs were retrospectively analyzed in institutional review board–approved studies (H-41604 and H-43439).
Generation of mLST products
mLSTs were generated as previously described.10 In brief, monocyte-derived dendritic cells were generated from donor peripheral blood, loaded with peptide mixtures (pepmixes, ie, panels of 15-mer peptides overlapping by 11 amino acids) spanning Survivin, WT1, PRAME, and NY-ESO-1 (JPT Peptide Technologies, Berlin, Germany) and cocultured with peripheral blood mononuclear cells (PBMCs) in the presence of a Th1-polarizing cytokine cocktail [IL7 (10 ng/mL), IL12 (10 ng/mL), IL15 (5 ng/mL), and IL6 (10 ng/mL)]. From day 10, responder T cells were restimulated weekly with pepmix-pulsed DCs in the presence of IL15 (5 ng/mL) or IL2 (50-100 U/mL) until sufficient numbers were achieved for patient infusion and requisite release testing.
mLST characterization and immune monitoring
Full details are found in supplemental Methods. In brief, an enzyme-linked immunospot (ELIspot) assay and immunophenotyping, intracellular cytokine staining (ICS), and 51chromium release cytotoxicity analyses were performed on aliquots of mLSTs (based on availability of material).
Tumor antigen profiling
Preinfusion and/or pre-HCT AML/MDS tissue blocks or slides were obtained when available (on protocol H-15280). Immunohistochemistry (IHC) was performed with a 1-step staining technique. In brief, formalin-fixed, paraffin-embedded, positively charged, unstained slides of tumor biopsy specimens (or, where applicable, clot sections) were obtained and underwent IHC staining (supplemental Methods).
Statistical analysis
Descriptive statistics were calculated to summarize clinical characteristics using mean, standard deviation (SD), standard error of the mean (SEM), median, and range. Dose escalation was applied independently and concurrently within the 2 arms (adjuvant and active disease), using the modified continual reassessment method (see “Study Design” in the full clinical protocol supplied in the supplemental Materials) to determine the maximum tolerated dose (MTD) of mLSTs, with MTD, defined as the highest dose level at which the probability of dose-limiting toxicity (DLT) was 20%, at most. Patients could be enrolled more than once to receive additional T-cell infusions if they did not qualify to receive additional cells per protocol, and the investigator(s) believed they could benefit from additional infusions, after appropriate regulatory approvals. However, per continual reassessment method each patient was counted only once (at the highest DL) for dose escalation calculations. A DLT was defined as grade 3 to 4 GVHD or National Cancer Institute Common Toxicity Criteria grade 3 to 5 within 4 weeks of infusion of mLSTs. Leukemia-free survival and overall survival (LFS and OS) were calculated from the time of the first mLST infusion to the date of relapse or death or were censored at last follow-up. The data cutoff date for analysis was 5 August 2020. Survival curves and median survival times were estimated by the Kaplan-Meier method. The rates of relapses between groups were compared by using Fisher’s exact test. Graphs were compiled with GraphPad Prism 6.
Results
Patients
Twenty-nine patients with a diagnosis of AML or MDS, who underwent HCT and whose donors provided PBMCs, were eligible, and we were able to generate mLSTs for infusion for all. Two donors had to undergo 2 separate procurements to achieve predetermined cell doses for infusion. Thus, a total of 31 products were generated from 29 donors. Nine patients were not infused, whereas 20 patients (17 adults and 3 children), with 5 patients enrolled and treated twice, representing a total of 25 enrollments, were administered mLSTs in the study. See CONSORT diagram in supplemental Figure 1 for details.
Adjuvant arm.
Seventeen enrollees (including patients 6 and 9, who were enrolled and treated again after receiving additional therapy for relapse) received mLSTs while in CR (adjuvant group; referral patterns listed in supplemental Results). The characteristics of treated patients are detailed in Table 1. Patients referred to the adjuvant arm of this trial were enriched for high risk of post-HCT relapse because they had 1 or more of the following characteristics: a history of post-HCT relapse (n = 5), induction failure or ≥CR2 before HCT (n = 7), and/or high-risk molecular features (n = 12; Table 1; supplemental Table 1). The median post-HCT time to mLST infusion was 117 days, a time frame that was driven primarily by the time needed to procure donors and manufacture mLSTs (details in supplemental Results).
Characteristics of patients treated in the adjuvant arm
| Never relapsed after HCT | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Donor | DL | Age/sex | Adverse-risk features | Prior treatments | Time to T cells after HCT (in days) | Donor chimerisms (blood or marrow), % | Ongoing tacrolimus on day of infusion |
| 1 | MRD | 1 | 57/F | FLT3-ITD | CIA→sorafenib→CIAx2→RIC-HCT | 117 | 100 | Yes |
| 2 | MRD | 1 | 18/F | FLT3-ITD | AAML 1031 (arm C-sorafenib)→MAC-HCT | 155 | 90 | Yes |
| 3 | MRD | 1 | 55/F | MLL-r | 7+3→HiDAC→MAC-HCT | 76 | 100 | Yes |
| 5 | MRD | 2 | 53/F | DNMT3A mut | 7+3→HiDAC→MAC-HCT | 63 | 100 | Yes |
| 8 | MRD | 2 | 65/M | MLL-r | 7+3×2→5-Azax11→RIC-HCT | 156 | 100 | Yes |
| 9 | MRD | 3 | 45/M | Ph+AML | 7+3+Imatinib→MAC-HCT | 106 | 100 | yes |
| 10 | MRD | 3 | 51/F | AML CR2 | 7+3→HiDAC→relapse→FLA→HiDAC→MAC-HCT | 106 | 100 | Yes |
| 4 | MRD | 1 | 54/F | Complex-rIPSS: Int-2 | 5-Azax11→transf-dep→RIC-SCT | 66 | 100 | Yes |
| 11 | MRD | 3 | 53/F | CR2 (MRD+ at HCT) | 7+3→HiDAC→relapse→FLA→ MRD+→MAC-HCT | 112 | 100 | None |
| 14 | MRD | 3 | 18/F | FLT3-ITD (MRD+ at HCT) | AAML1031→relapse→CPX-351→FLA(G)→Ara-C/peg/midostaurin→refractory→venetoclax/decitabine→MRD+→MAC-HCT | 132 | 100 | Unknown |
| 13 | MRD | 3 | 26/M | TP53 mut MDS-EB2 | 5-aza+ venetoclax→MAC-HCT | 132 | 100 | Yes |
| 15 | MUD | 5 | 67/M | CMML→AMML (PIF) | 7+3→residual disease→venetoclax+5-aza→RIC-HCT | 230 | 91 | None |
| Never relapsed after HCT | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Donor | DL | Age/sex | Adverse-risk features | Prior treatments | Time to T cells after HCT (in days) | Donor chimerisms (blood or marrow), % | Ongoing tacrolimus on day of infusion |
| 1 | MRD | 1 | 57/F | FLT3-ITD | CIA→sorafenib→CIAx2→RIC-HCT | 117 | 100 | Yes |
| 2 | MRD | 1 | 18/F | FLT3-ITD | AAML 1031 (arm C-sorafenib)→MAC-HCT | 155 | 90 | Yes |
| 3 | MRD | 1 | 55/F | MLL-r | 7+3→HiDAC→MAC-HCT | 76 | 100 | Yes |
| 5 | MRD | 2 | 53/F | DNMT3A mut | 7+3→HiDAC→MAC-HCT | 63 | 100 | Yes |
| 8 | MRD | 2 | 65/M | MLL-r | 7+3×2→5-Azax11→RIC-HCT | 156 | 100 | Yes |
| 9 | MRD | 3 | 45/M | Ph+AML | 7+3+Imatinib→MAC-HCT | 106 | 100 | yes |
| 10 | MRD | 3 | 51/F | AML CR2 | 7+3→HiDAC→relapse→FLA→HiDAC→MAC-HCT | 106 | 100 | Yes |
| 4 | MRD | 1 | 54/F | Complex-rIPSS: Int-2 | 5-Azax11→transf-dep→RIC-SCT | 66 | 100 | Yes |
| 11 | MRD | 3 | 53/F | CR2 (MRD+ at HCT) | 7+3→HiDAC→relapse→FLA→ MRD+→MAC-HCT | 112 | 100 | None |
| 14 | MRD | 3 | 18/F | FLT3-ITD (MRD+ at HCT) | AAML1031→relapse→CPX-351→FLA(G)→Ara-C/peg/midostaurin→refractory→venetoclax/decitabine→MRD+→MAC-HCT | 132 | 100 | Unknown |
| 13 | MRD | 3 | 26/M | TP53 mut MDS-EB2 | 5-aza+ venetoclax→MAC-HCT | 132 | 100 | Yes |
| 15 | MUD | 5 | 67/M | CMML→AMML (PIF) | 7+3→residual disease→venetoclax+5-aza→RIC-HCT | 230 | 91 | None |
| Relapsed after HCT, but in CR after salvage therapy | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Donor | DL | Age/sex | Adverse-risk features | Prior treatments | Time to relapse after HCT (in days) | Donor chimerisms (blood or marrow), % | Ongoing tacrolimus on day of infusion |
| 6 | MRD | 2 | 70/F | AML CR3 | 7+3→HiDAC→CIA→RIC-HCT-relapse→7+3 | 800 | 100 | None |
| 12 | MRD | 3 | 55/M | Ph+, t-AML | 7+3→RIC-HCT→relapse→7+3 | 2130 | 100 | None |
| 7 | MRD | 2 | 58/M | RAEB-1 rIPSS: Int-2→t-AML in CR2 | Decitabine→RIC-HCT→relapse with RAEB→CIA→relapse as MDS→DLI (×4) | 356 | 100 | None |
| 9 | MRD | 4 | 47/M | Ph+AML in CR2 | 7+3+Imatinib→MAC-HCT→mLST→molecular relapse→decitabine-dasatinib | 460 | 100 | None |
| 6 | MRD | 4 | 73/F | AML CR5 | 7+3→HiDAC→IA→RIC-HCT-relapse→7+3→mLST→relapse→IT chemo and XRT→relapse→IT chemo and XRT | 1330 | 100 | None |
| Relapsed after HCT, but in CR after salvage therapy | ||||||||
|---|---|---|---|---|---|---|---|---|
| ID | Donor | DL | Age/sex | Adverse-risk features | Prior treatments | Time to relapse after HCT (in days) | Donor chimerisms (blood or marrow), % | Ongoing tacrolimus on day of infusion |
| 6 | MRD | 2 | 70/F | AML CR3 | 7+3→HiDAC→CIA→RIC-HCT-relapse→7+3 | 800 | 100 | None |
| 12 | MRD | 3 | 55/M | Ph+, t-AML | 7+3→RIC-HCT→relapse→7+3 | 2130 | 100 | None |
| 7 | MRD | 2 | 58/M | RAEB-1 rIPSS: Int-2→t-AML in CR2 | Decitabine→RIC-HCT→relapse with RAEB→CIA→relapse as MDS→DLI (×4) | 356 | 100 | None |
| 9 | MRD | 4 | 47/M | Ph+AML in CR2 | 7+3+Imatinib→MAC-HCT→mLST→molecular relapse→decitabine-dasatinib | 460 | 100 | None |
| 6 | MRD | 4 | 73/F | AML CR5 | 7+3→HiDAC→IA→RIC-HCT-relapse→7+3→mLST→relapse→IT chemo and XRT→relapse→IT chemo and XRT | 1330 | 100 | None |
All patients were in morphological CR and with no detectable disease on preinfusion flow cytometry or genetic analysis. Bold, italicized text highlights major events during the course of the patient's disease.
5-aza, 5 azacytidine; 7+3, 7 d of cytarabine infusion and 3 d of idarubicin; AAML 1031, up-front pediatric treatment protocol; Ara-C, cytarabine; CIA, clofarabine, idarubicin, and cytarabine; CMML→AMML, acute converted from chronic myelomonocytic leukemia; DNMT3A mut, DNA methyltransferase, 3A mutation; F, female; FLA(G), fludarabine and cytarabine with G-CSF; FLT3-ITD, fms-like tyrosine kinase 3 receptor1-internal tandem duplication; HiDAC, high dose (>1 g/m2 cytarabine), ID, patient ID number; Int-2, intermediate-2; IT chemo, intrathecal chemotherapy only; M, male; MAC, fully myeloablative pre-HCT conditioning chemotherapy; MDS-EB2, myelodysplastic syndrome-excess blasts-2; MLL-r, mixed lineage leukemia-1 gene rearrangement; MRD+, measurable residual disease present; Peg, pegasparaginase; Ph+, Philadelphia chromosome–positive (BCR-ABL rearranged); PIF, primary induction failure; RAEB, refractory anemia with excess of blasts; RIC, reduced-intensity pre-HCT conditioning chemotherapy; rIPSS, revised International Prognostic System for MDS; t-AML, therapy-related AML; TP53 mut, mutated TP53 gene, XRT, radiotherapy.
Active disease arm.
All 8 enrollees in the active disease cohort (patient 1 crossed over to the active arm and was reenrolled as patient A1 after US Food and Drug Administration approval), whereas patients A5 and A6 were enrolled twice. All 3 had active AML for which another line of salvage therapy or HCT had failed after their first mLST infusion, so were reenrolled. Patients in this cohort received a median of 5 prior lines of therapy (range, 4-10; Table 2). All had measurable active leukemia at the time of mLST infusion, with a median of 30% (30%-70%) marrow blasts before infusion.
Characteristics of patients treated on the active disease arm, with any measurable disease before infusion
| ID | Donor | DL | Age/sex | Disease | Prior treatments | Donor chimerisms (blood or marrow) | Ongoing tacrolimus on day of infusion |
|---|---|---|---|---|---|---|---|
| A2 | MRD | 1 | 70/M | IDH1mut | 7+3→decitabine→IDH inhibitor→cutis relapse→CIA→RIC-HCT→relapse | 100% (skin relapse) | No |
| A3 | Haplo | 1 | 16/M | MDS→AML | Double cord HCT→AML relapse→C→haplo-HCT×2→relapse | <20% | No |
| 1 & A1* | MRD | 1 | 57/F | FLT3-ITD | CIA→sorafenib→CIA×2→RIC-HCT→mLST→steroids→relapse | 100% (bone relapse) | No |
| A4 | MRD | 2 | 55/M | PIF | 7+3→HiDAC×4→RIC-HCT→relapse→DLI×4→MEC→5-aza→relapse | Not checked | No |
| A5* | Haplo | 2 | 23/M | Del 17p | CIA×3→haplo-HCT→relapse→CIA-decitabine→haplo-HCT→5-aza→nivolumab→CD123 BiTE→MEC-decitabine→ midostaurin→relapse | Not checked | No |
| A5* | Haplo | 2 | 23/M | Del 17p | CIA×3→ haplo-HCT#1→relapse→ CIA-decitabine→haplo-HCT#2→5-aza→nivolumab→CD123 BiTE→MEC-decitabine→midostaurin→relapse→mLST→haplo-HCT#3→relapse | Not checked | No |
| A6* | MRD | 3 | 20/F | FLT3-ITD | 7+3→HiDAC→MAC-HCT→relapse→CIA→relapse | 45% | No |
| A6* | MRD | 3 | 20/F | FLT3-ITD | 7+3→HiDAC→MAC-HCT→relapse→CIA→relapse→mLST→CIA+decitabine→mLST | Not checked | No |
| ID | Donor | DL | Age/sex | Disease | Prior treatments | Donor chimerisms (blood or marrow) | Ongoing tacrolimus on day of infusion |
|---|---|---|---|---|---|---|---|
| A2 | MRD | 1 | 70/M | IDH1mut | 7+3→decitabine→IDH inhibitor→cutis relapse→CIA→RIC-HCT→relapse | 100% (skin relapse) | No |
| A3 | Haplo | 1 | 16/M | MDS→AML | Double cord HCT→AML relapse→C→haplo-HCT×2→relapse | <20% | No |
| 1 & A1* | MRD | 1 | 57/F | FLT3-ITD | CIA→sorafenib→CIA×2→RIC-HCT→mLST→steroids→relapse | 100% (bone relapse) | No |
| A4 | MRD | 2 | 55/M | PIF | 7+3→HiDAC×4→RIC-HCT→relapse→DLI×4→MEC→5-aza→relapse | Not checked | No |
| A5* | Haplo | 2 | 23/M | Del 17p | CIA×3→haplo-HCT→relapse→CIA-decitabine→haplo-HCT→5-aza→nivolumab→CD123 BiTE→MEC-decitabine→ midostaurin→relapse | Not checked | No |
| A5* | Haplo | 2 | 23/M | Del 17p | CIA×3→ haplo-HCT#1→relapse→ CIA-decitabine→haplo-HCT#2→5-aza→nivolumab→CD123 BiTE→MEC-decitabine→midostaurin→relapse→mLST→haplo-HCT#3→relapse | Not checked | No |
| A6* | MRD | 3 | 20/F | FLT3-ITD | 7+3→HiDAC→MAC-HCT→relapse→CIA→relapse | 45% | No |
| A6* | MRD | 3 | 20/F | FLT3-ITD | 7+3→HiDAC→MAC-HCT→relapse→CIA→relapse→mLST→CIA+decitabine→mLST | Not checked | No |
5-aza, 5 azacytidine; 7+3, 7 d of cytarabine infusion and 3 d of idarubicin; CD123 BiTE, CD123-CD3 bispecific T-cell engager on an investigational protocol; CIA, clofarabine, idarubicin, and cytarabine; del 17p, deletion of short arm of chromosome 17; F, female; FLT3-ITD, fms-like tyrosine kinase 3 receptor-internal tandem duplication; Haplo, haploidentical; HiDAC, high dose (>1 g/m2 cytarabine); ID, patient ID number; IDHmut, IDH mutation; M, male; MAC, fully myeloablative pre-HCT conditioning chemotherapy; MEC, mitoxantrone, etoposide, and cytarabine; PIF, primary induction failure; RIC, reduced-intensity pre-HCT conditioning chemotherapy.
Reenrolled into the active arm after relapse.
mLST characterization
Tumor-specific T cells are present in the circulation of healthy individuals, including HSC donors, but at low levels. Indeed, the mean frequency of circulating T cells that were reactive against WT1, PRAME, NY-ESO-1, and Survivin in our donors was just 2 ± 1 spot-forming cells (SFCs)/5 × 105 PBMCs for each antigen, as determined by IFN-γ ELIspot assay. Thus, to maximize therapeutic benefit and minimize the risk of GVHD from alloreactive T cells, we selectively activated and expanded the leukemia-reactive population using our previously described methodology.10 After 34 ± 7 days in culture and 2 to 4 rounds of in vitro stimulation, we achieved a mean 16 ± 2-fold increase in cells that were enriched for tumor-reactive populations. Of the 31 lines that were generated, the mean frequency of mLSTs was 210 ± 58 SFCs per 2 × 105, with PRAME inducing the strongest activity (114 ± 35 SFCs per 2 × 105 cells), followed in descending order by WT1 (53 ± 23 SFCs), NY-ESO-1 (34 ± 11 SFCs), and Survivin (10 ± 3 SFCs) (Figure 1B). These expanded cells exhibited no alloreactivity against patient-derived, nonmalignant phytohemagglutinin (PHA) induced blasts (mean, 3.0% ± 0.5% specific lysis; effector/target [E/T], 20:1; n = 31). Of note, <10% specific lysis of patient-derived PHA blasts at an E/T of 20:1 was set as the safety release criterion and was met by all products (Figure 1C). In addition, these mLSTs killed antigen-loaded targets (mean, 24% ± 7% specific lysis, E/T 20:1, n = 11, in whom sufficient residual material was available; Figure 1D). The expanded mLSTs were polyclonal as assessed by both TCR-vβ deep sequencing (Figure 1E) and phenotypic analysis (Figure 1F), with a mixture of CD4+ (32.4% ± 4.5%) and CD8+ (44.6% ± 3.9%) T cells that were activated (CD3+/CD69+, 35.1% ± 2.7%) and expressed central (CD45RO+/CD62L+, 16.6% ± 2.5%) and effector memory markers (CD45RO+/CD62L−, 50.8% ± 3.4%). Lines with >50% CD4+ cells (n = 7) did not demonstrate any statistically significant differences in magnitude of tumor-associated antigen (TAA) specificity or killing capacity when compared with those that had <50% CD4+ cells (n = 20).
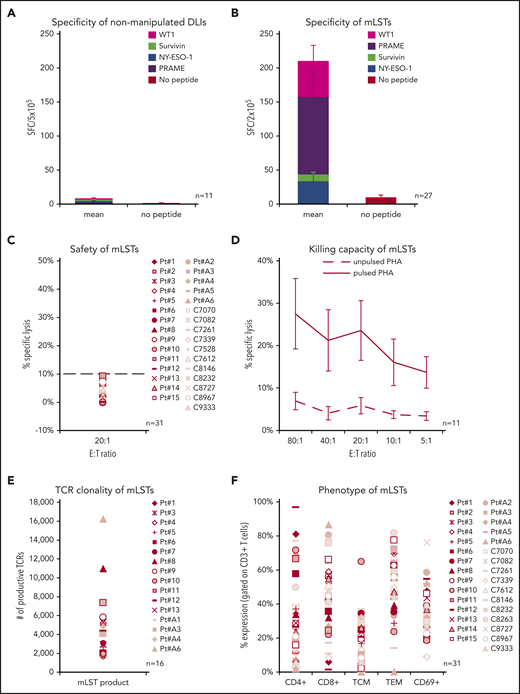
In vitro characteristics of mLSTs. Leukemia TAA-directed activity of nonmanipulated donor lymphocytes (A) and ex vivo expanded mLSTs (B). Mean ± SEM. mLSTs did not kill normal recipient cells (C), but killed TAA-pulsed normal cells tested at E/T ratios from 80:1 to 5:1 (D). Mean ± SEM. Polyclonality of mLSTs, as assessed by TCR-vβ deep sequencing (E) and immunophenotyping (F). (C,E-F) Each symbol represents an individual product.
In vitro characteristics of mLSTs. Leukemia TAA-directed activity of nonmanipulated donor lymphocytes (A) and ex vivo expanded mLSTs (B). Mean ± SEM. mLSTs did not kill normal recipient cells (C), but killed TAA-pulsed normal cells tested at E/T ratios from 80:1 to 5:1 (D). Mean ± SEM. Polyclonality of mLSTs, as assessed by TCR-vβ deep sequencing (E) and immunophenotyping (F). (C,E-F) Each symbol represents an individual product.
In vivo safety of mLSTs
Forty-eight infusions were administered to 25 enrollees across both arms, and only 3 (12% of all enrollees) developed de novo (grade 1) or worsening (grade 2) acute postinfusion GVHD, and 4 (16% of all enrollees) developed de novo mild chronic GVHD (Table 3). Only 1 patient (with grade 2 upper gastrointestinal [GI] GVHD) required systemic steroid treatment. Importantly, there were no cases of cytokine release syndrome, neurotoxicity, or persistent myelotoxicity (>28 days). Hepatitis (characterized by aspartate and alanine transaminase [ALT/AST] elevations only) was deemed treatment-related because of its occurrence in 6 patients (who received infusions at the 3 lowest DLs) and temporally associated with infusions. Except for 1 case of grade 3 hepatitis treated with systemic steroids, all others were grade ≤2 and self-limiting (Table 4). No liver biopsies were performed to confirm or refute atypical GVHD as a cause of hepatitis.
Adverse events of special interest
| Acute GVHD | ||||||
|---|---|---|---|---|---|---|
| DL | Organ | Enrollees, n (%) | Onset in relation to infusion | Max overall grade | Systemic steroids | Outcome |
| DL2 and DL5 | Skin | 2 of 24 (8) | 7 d after infusion for both | 1 | No | Resolved in 1 wk for both |
| DL2 | Upper GI | 1 of 24 (4) | 14 d after infusion | 2 | Yes | Resolved in 2 wk, steroid treatment discontinued |
| Acute GVHD | ||||||
|---|---|---|---|---|---|---|
| DL | Organ | Enrollees, n (%) | Onset in relation to infusion | Max overall grade | Systemic steroids | Outcome |
| DL2 and DL5 | Skin | 2 of 24 (8) | 7 d after infusion for both | 1 | No | Resolved in 1 wk for both |
| DL2 | Upper GI | 1 of 24 (4) | 14 d after infusion | 2 | Yes | Resolved in 2 wk, steroid treatment discontinued |
| Chronic GVHD | ||||||
|---|---|---|---|---|---|---|
| DL | Organ | Enrollees, n (%) | Onset in relation to infusion | Max overall severity | Systemic treatment | Outcome |
| DL1 | Vaginal | 1 of 24 (4) | 1 y after infusion | Mild | No | Ongoing |
| DL3 | Skin | 1 of 24 (4) | 9 mo after infusion | Mild | No | Ongoing |
| DL3 | Joint | 1 of 24 (4) | 9 mo after infusion | Mild | No | Ongoing |
| DL3 | Eyes | 1 of 24 (4) | 9 mo after infusion | Mild | No | Ongoing |
| CRS | None at any dose level | |||||
| Neurotoxicity | None at any dose level | |||||
| Myelotoxicity (cytopenias persisting >28 d) | None at any dose level | |||||
| Chronic GVHD | ||||||
|---|---|---|---|---|---|---|
| DL | Organ | Enrollees, n (%) | Onset in relation to infusion | Max overall severity | Systemic treatment | Outcome |
| DL1 | Vaginal | 1 of 24 (4) | 1 y after infusion | Mild | No | Ongoing |
| DL3 | Skin | 1 of 24 (4) | 9 mo after infusion | Mild | No | Ongoing |
| DL3 | Joint | 1 of 24 (4) | 9 mo after infusion | Mild | No | Ongoing |
| DL3 | Eyes | 1 of 24 (4) | 9 mo after infusion | Mild | No | Ongoing |
| CRS | None at any dose level | |||||
| Neurotoxicity | None at any dose level | |||||
| Myelotoxicity (cytopenias persisting >28 d) | None at any dose level | |||||
CRS, cytokine release syndrome; Max, maximum grade seen in any of the patients at the indicated dose level.
All other treatment-related adverse events
| Incident | Enrollees, n | Onset in relation to infusion | Max grade | Treatment, if any | Status |
|---|---|---|---|---|---|
| DL1 | |||||
| Hepatitis | 1 | 7 d | 3 | Prednisone 0.5 mg/kg | Resolved 54 d later |
| Nausea/vomiting | 3 | 5 d, 2 d, and 1 mo | 1 | None | Resolved in 7 d |
| Skin dryness | 1 | 7 d | 1 | None | Resolved in 7 d |
| Incident | Enrollees, n | Onset in relation to infusion | Max grade | Treatment, if any | Status |
|---|---|---|---|---|---|
| DL1 | |||||
| Hepatitis | 1 | 7 d | 3 | Prednisone 0.5 mg/kg | Resolved 54 d later |
| Nausea/vomiting | 3 | 5 d, 2 d, and 1 mo | 1 | None | Resolved in 7 d |
| Skin dryness | 1 | 7 d | 1 | None | Resolved in 7 d |
| Incident | Patients, n | Onset in relation to infusion | Max grade | Treatment, if any | Status |
|---|---|---|---|---|---|
| DL2 | |||||
| Dry eyes | 2 | 7 d and 9 d | 1 | Topical treatments | Stable, Schirmer test not done |
| Hepatitis | 1 | 26 d | 2 | Coincident with grade 2 aGVHD above prednisone 0.5 mg/kg | Resolved in 2 wk |
| Anemia | 1 | 32 d | 1 | Coincident with grade 2 aGVHD above prednisone 0.5 mg/kg | Resolved in 7 d |
| Fatigue | 1 | 27 d | 2 | Coincident with RSV infection | Resolved in 7 d |
| Anorexia | 1 | 27 d | 2 | Coincident with RSV infection | Resolved in 7 d |
| Dizziness | 1 | 9 d | 1 | None | Resolved in 7 d |
| DL3 | |||||
| Diarrhea | 1 | 3 d | 1 | None | Resolved in 1 d |
| Fatigue | 2 | 7 d and 14 d | 1 | None | Resolved by day 28 |
| Hepatitis | 4 | 7 d, 7 d, 5 d, and 26 d | 1 | None | Resolved by day 60 in all |
| DL4 | |||||
| Diarrhea | 1 | 14 d | 1 | None | Resolved in 7 d |
| Lymphocytosis | 1 | 28 d | 2 | None | Ongoing without symptoms |
| DL5 | None | ||||
| Incident | Patients, n | Onset in relation to infusion | Max grade | Treatment, if any | Status |
|---|---|---|---|---|---|
| DL2 | |||||
| Dry eyes | 2 | 7 d and 9 d | 1 | Topical treatments | Stable, Schirmer test not done |
| Hepatitis | 1 | 26 d | 2 | Coincident with grade 2 aGVHD above prednisone 0.5 mg/kg | Resolved in 2 wk |
| Anemia | 1 | 32 d | 1 | Coincident with grade 2 aGVHD above prednisone 0.5 mg/kg | Resolved in 7 d |
| Fatigue | 1 | 27 d | 2 | Coincident with RSV infection | Resolved in 7 d |
| Anorexia | 1 | 27 d | 2 | Coincident with RSV infection | Resolved in 7 d |
| Dizziness | 1 | 9 d | 1 | None | Resolved in 7 d |
| DL3 | |||||
| Diarrhea | 1 | 3 d | 1 | None | Resolved in 1 d |
| Fatigue | 2 | 7 d and 14 d | 1 | None | Resolved by day 28 |
| Hepatitis | 4 | 7 d, 7 d, 5 d, and 26 d | 1 | None | Resolved by day 60 in all |
| DL4 | |||||
| Diarrhea | 1 | 14 d | 1 | None | Resolved in 7 d |
| Lymphocytosis | 1 | 28 d | 2 | None | Ongoing without symptoms |
| DL5 | None | ||||
Max, maximum grade seen in any of the patients at the indicated dose level; RSV, respiratory syncytial virus.
Outside of the aforementioned events, there were no other grade ≥3 adverse events noted on the adjuvant arm of the study. Of the 8 enrollees treated on the active arm of the study, all had 1 or more grade ≥3 cytopenias (anemia, leukopenia, or thrombocytopenia) related to their active AML/MDS, with no or only transient worsening after mLST infusion; all recovered to baseline by postinfusion day 28.
Adjuvant arm outcomes
Seventeen enrollees with high-risk AML/MDS received infusions of mLSTs; 5 recipients had experienced post-HCT relapse but achieved a CR with subsequent salvage regimens before mLST infusion (Table 1). Six of 17 enrollees relapsed (Figure 2) at a median of 9.5 months (range, 5-12.3 months) after infusion. Notably, 1 patient had only molecular relapse without detectable blasts on marrow pathology; 3 other relapses occurred in immune-privileged extramedullary sites without bone marrow involvement, although all 3 initially had marrow-only disease. Two patients with central nervous system–only (CNS) relapse received intrathecal chemotherapy and craniospinal radiation therapy with complete resolution of disease; patient 6 was subsequently reenrolled and received mLSTs at a higher dose level (DL4). At this writing, both patients are alive and in CR >3 years since their initial mLST infusion. Decitabine salvage therapy failed in patient 1, who experienced multiple foci of relapse within vertebral bodies (4 cortical bony sites), without involvement of the marrow, but achieved a CR after transitioning to the mLST active disease arm (as patient A1) and receiving an additional dose of cells. Patient 9, who experienced only molecular relapse, was treated with 4 cycles of decitabine with dasatinib, after which he achieved a molecular CR. He also reenrolled on the adjuvant arm of the trial and received another infusion of mLSTs at a higher dose level (DL4). Finally, the remaining 2 patients with frank marrow relapse went on to receive salvage systemic chemotherapy, but neither responded. Overall, 11 of 17 enrollees never relapsed after mLST infusion (median LFS not reached at a median follow-up of 1.9 years), and 11 of 15 patients remained alive (estimated 2-year OS of 77%; supplemental Figure 2) at a median follow-up of 1.9 years after infusion (range, 6-51 months; Figure 2), which compares favorably with HCT outcomes for risk-matched patients with AML/MDS after HCT (median LFS of 9-15 months and 2-year survival probability of 42% [38%-46%])11,12 (Figure 2B).
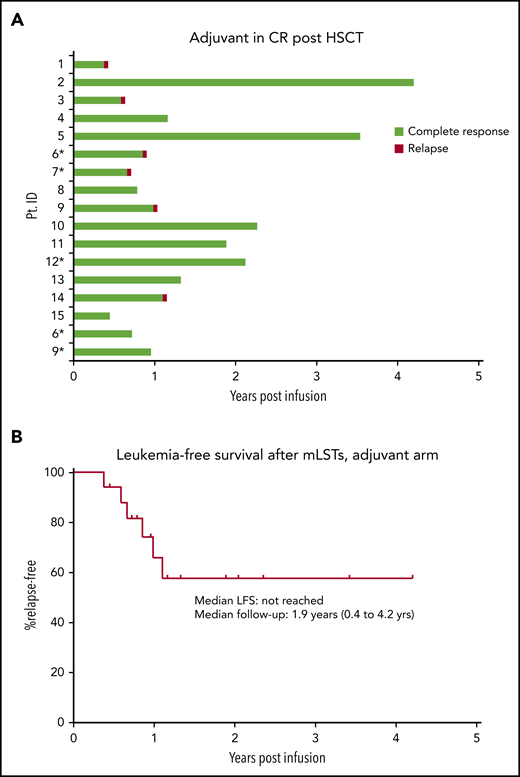
Clinical outcomes: adjuvant arm. (A) Swimmer plots depicting outcomes after infusion of mLSTs in patients with AML/MDS who were in remission at the time of infusion. *Individuals who relapsed after HCT but were in morphological remission at the time of T-cell infusion. (B) Kaplan-Meier estimates of LFS in the adjuvant group.
Clinical outcomes: adjuvant arm. (A) Swimmer plots depicting outcomes after infusion of mLSTs in patients with AML/MDS who were in remission at the time of infusion. *Individuals who relapsed after HCT but were in morphological remission at the time of T-cell infusion. (B) Kaplan-Meier estimates of LFS in the adjuvant group.
Evidence of immune escape after mLST infusion
Of 6 patients who relapsed after mLST infusion, 5 had available specimens that enabled correlative studies to assess the mechanism of disease relapse (the sixth had evidence of molecular disease only). All 5 demonstrated 1 or more known mechanisms of immune escape (Figure 3A); upregulation of PD-L113 in 3 of 5 (Figure 3B), along with tumor relapse in immune-privileged sites (CNS and bone, compared with marrow disease at initial treatment) in 3 of 6 (Figure 3C),14-16 decreased major histocompatibility complex class 2 expression in 4 of 5 (HLA-DRdim/loss at relapse, compared with the preinfusion or diagnosis sample, as assessed by flow cytometry; Figure 3D) and/or loss of target antigen expression in 3 of 5 (Figure 3E).13,17,18 Of 21 consecutive patients with AML/MDS who underwent matched sibling donor-HCT or DLI at our center, compared with 12 of those patients who experienced post-HCT relapses but who did not receive mLSTs, only 1 patient experienced extramedullary relapse (P = .09; ns), and 1 of 9 (of 12 in whom HLA-DR status was known) had HLA-DRdim/loss at relapse (P = .023; supplemental Table 1).
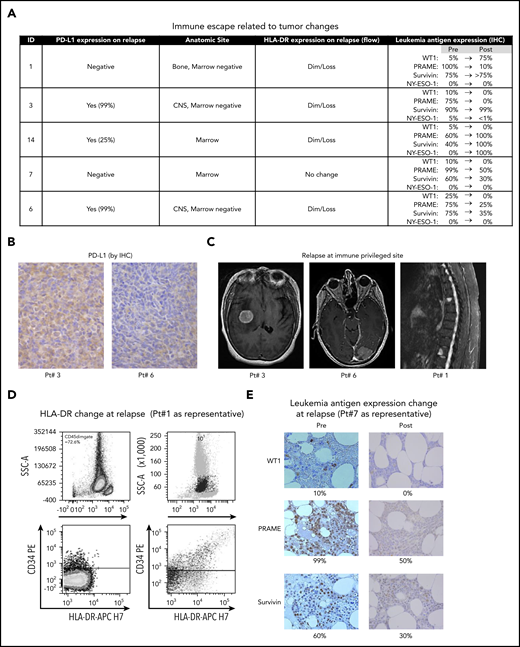
Mechanisms of immune escape at relapse. (A) Summary of mechanisms of immune escape observed in each relapsing patient. (B) Expression of PD-L1 as estimated by IHC in relapsing tumors obtained from patients 3 and 6. Original magnification ×400. (C) MRI imaging in 3 individuals demonstrating extramedullary (immune-privileged anatomic site) relapses. (D) Representative flow cytometry plot from patient 1 depicting loss of HLA-DR expression at relapse. (E) Downregulation of expression of target TAAs (WT1, PRAME, Survivin) on relapsing AML/MDS cells in patient 7. IHC; original magnification, ×400.
Mechanisms of immune escape at relapse. (A) Summary of mechanisms of immune escape observed in each relapsing patient. (B) Expression of PD-L1 as estimated by IHC in relapsing tumors obtained from patients 3 and 6. Original magnification ×400. (C) MRI imaging in 3 individuals demonstrating extramedullary (immune-privileged anatomic site) relapses. (D) Representative flow cytometry plot from patient 1 depicting loss of HLA-DR expression at relapse. (E) Downregulation of expression of target TAAs (WT1, PRAME, Survivin) on relapsing AML/MDS cells in patient 7. IHC; original magnification, ×400.
In patients treated with mLSTs the frequency of infused cells, as measured by TCR-vβ clone tracking (supplemental Figure 3), appeared to decline in those who relapsed compared with those who did not, although the small samples prohibited statistical comparisons. However, at relapse, the frequency of functional TAA-reactive T cells (IFN-γ ELIspot) was markedly lower than it was early after infusion (supplemental Figure 4). Notably, no significant differences were seen in proportions of CD4+, CD8+, PD1+, LAG+, central or effector memory subsets in mLST products administered to those who relapsed vs those who remained in CR. Of those who relapsed, 3 were reenrolled and successfully re-treated with mLSTs (1 on the active disease arm and 2 on the adjuvant arm) after potential mechanisms of immune escape were addressed (see “Adjuvant arm outcomes” for details). All 3 remained in CR after retreatment for at least as long or longer (>1 year in 2 cases, ongoing) than after the initial mLST infusion.
Direct anti-AML/MDS effects
To demonstrate direct antitumor effects of mLSTs, 8 enrollees with HCT-resistant AML/MDS were treated with mLSTs. Table 2 demonstrates that all 8 had relapsed after HCT with disease that was resistant to salvage measures. Two achieved an objective response after infusion of mLSTs (Figure 4). In those who did not respond to therapy, we saw a decline in the expression of target TAAs on paired pre- and posttreatment biopsy samples (n = 3; supplemental Table 2; supplemental Figures 5A and 6A) and loss or downregulation of HLA-DR (1 of 4 with known HLA-DR status; supplemental Table 2). Circulating frequencies of TAA-specific T cells declined coincident with declining TAA expression on progressing tumors (supplemental Figures 5B and 6B).
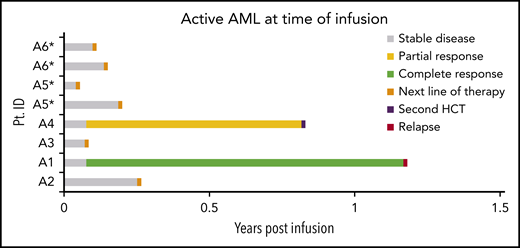
Clinical outcomes: active disease arm. Swimmer plots depicting outcomes after infusion in patients with AML/MDS who had refractory disease at the time of infusion.
Clinical outcomes: active disease arm. Swimmer plots depicting outcomes after infusion in patients with AML/MDS who had refractory disease at the time of infusion.
Patient 1 was initially infused in the adjuvant arm but relapsed with discrete bone lesions coincident with loss of mLST-derived T-cell clones after treatment with systemic steroids for grade 3 hepatitis. After relapse, the patient received hypomethylating therapy (decitabine), with no response, and was subsequently reenrolled on the active disease arm (as patient A1, at DL1) after discontinuation of systemic steroids. Within 4 weeks of a single infusion of cells, she entered CR (Figure 5A-B) and received 3 additional infusions (all at DL1) of mLSTs 4 to 6 weeks apart with no recurrence of hepatitis. Before the mLST infusions, biopsy specimens of a bone lesion demonstrated a dense CD3+ infiltrate surrounding CD33+ blasts indicative of present but inactive antileukemic T cells (Figure 5C). Concomitant with her achievement of CR, we detected an increase in both CD8+ and CD4+ T WT1-reactive T cells, as assessed by both IFN-γ ELISpot (on PBMCs; Figure 5D) and ICS (gated on CD3+ cells; Figure 5E), and derived from infused mLSTs (Figure 5F).
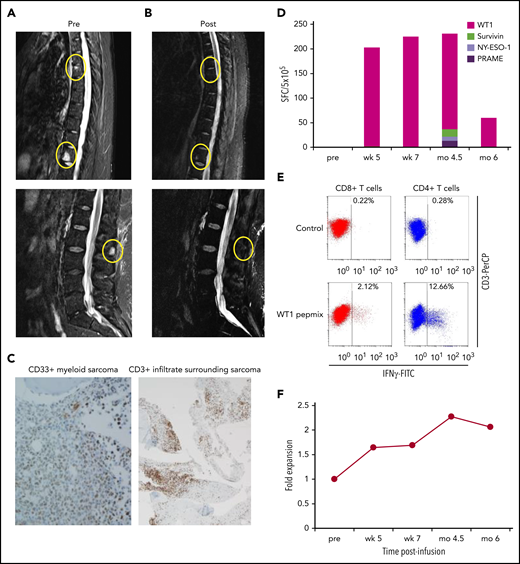
Complete remission from active AML after mLST infusion. MRI images of AML invading bone before (A) and after (B) mLST infusion. (C) Relapsed AML cells (CD33+ blasts; IHC). Hematoxylin and eosin; original magnification ×400. Cells were surrounded by a dense infiltrate of CD3+ T cells. IHC; original magnification ×10. Changes in frequency of circulating mLSTs after infusion, as measured by IFN-γ ELISpot (D) and polyclonality of circulating WT1-specific T cells, as estimated by ICS (E) performed on fresh PBMCs at the 6-month time point. (F) Change in the frequency of mLST-derived TCR clones represented as fold change from baseline in the repertoire frequency.
Complete remission from active AML after mLST infusion. MRI images of AML invading bone before (A) and after (B) mLST infusion. (C) Relapsed AML cells (CD33+ blasts; IHC). Hematoxylin and eosin; original magnification ×400. Cells were surrounded by a dense infiltrate of CD3+ T cells. IHC; original magnification ×10. Changes in frequency of circulating mLSTs after infusion, as measured by IFN-γ ELISpot (D) and polyclonality of circulating WT1-specific T cells, as estimated by ICS (E) performed on fresh PBMCs at the 6-month time point. (F) Change in the frequency of mLST-derived TCR clones represented as fold change from baseline in the repertoire frequency.
Similarly, patient A4, who had refractory AML, had a >50% reduction in marrow blast infiltration (from 40% before infusion to 15% by week 4 after infusion), concomitant with increasing blood counts (absolute neurophil count from 250 to >1000 by month 4 without growth factors), which ultimately enabled a second HCT. At the time of infusion, this patient’s blasts expressed WT1, PRAME, and Survivin and the observed clinical benefit occurred coincident with a detectable increase in the frequency of WT1-, PRAME-, and Survivin-reactive T cells. The expanded TAA-reactive T cells were 98% donor after infusion compared with 5% before infusion, indicating that they were of mLST origin; supplemental Figure 7).
Discussion
In this trial, we demonstrated the safety and activity of an allogeneic AML/MDS-targeted T-cell therapy. When administered to 25 enrollees at high risk or those who had relapsed after HCT, mLSTs at doses of 0.5 × 107 to 10 × 107 cells per square meter were well tolerated, with no cases of grade ≥3 acute GVHD (aGVHD) or any instance of extensive cGVHD. We observed antileukemia effects in both cohorts, evidenced by long post-HCT remissions in the adjuvant group (median LFS not reached at a median follow-up of 1.9 years, estimated 2-year OS of 77%) and objective responses (1 CR and 1 partial response) seen in the HCT-refractory, active disease cohort. Thus, outcomes from this trial demonstrate that mLST infusion may be a safe and effective alternative to DLI.
Prophylactic DLIs have been shown to reduce the incidence of relapse among those with high-risk AML (30% [DLI] vs >46% [no DLI] in one registry study).19 Unlike DLIs, which frequently induce severe GVHD (grade ≥3 in up to 30% of all recipients; range, 2-5) related to the concomitant presence of alloreactive T cells, we saw no grade ≥3 aGVHD and only 1 episode of grade 2 aGVHD in our 25 recipients, supporting the safety of mLSTs. In addition, we saw none of the toxicities associated with engineered T cells (eg, CAR T cells), including prolonged myelotoxicity, cytokine release syndrome, or neurotoxicity.20-22 One patient with an infusion of mLSTs at a cell dose of 0.5 × 107 cells per square meter, developed grade 3 hepatitis, and 5 others had transient grade ≤2 hepatitis after infusions at doses up to 2 × 107 cells per square meter, which showed an unexpected toxicity. Only patient 1 needed systemic corticosteroids (0.8 mg/kg prednisone) for the treatment of hepatitis, whereas patient 8, with grade 2 hepatitis, received 0.5 mg/kg prednisone to treat gastrointestinal GVHD. Both patients responded, and corticosteroids were eventually discontinued. Notably, patient 1 subsequently received 4 additional infusions of the same product at the same dose level (as patient A1) without recurrent hepatitis in the active disease arm. This result suggests that the initial event may have been a hepatitic pattern of acute GVHD, which is observed after transplantation23,24 as doses of calcineurin inhibitors are weaned. Thus, overall, mLSTs, infused at doses comparable with DLIs, were well tolerated and demonstrated a superior safety profile to DLIs and CAR-T cells. Standardization of donor procurements could ensure availability of mLSTs to all patients by day +30 after HCT in pivotal trials.
Prior attempts to harness the potential of donor-derived, leukemia-reactive T cells have mainly focused on targeting a single antigen or a single epitope restricted to 1 HLA type.25-29 Although effective at preventing relapse in select reports, these approaches have limited effectiveness in the treatment of frank post-HCT relapse. For example, Chapuis and colleagues administered donor-derived Epstein Barr virus–specific T cells engineered to express a TCR specific for an HLA-A2–restricted WT1 epitope in 11 patients with refractory AML and 12 patients in remission. Although treatment was restricted to HLA-A2+ individuals, patients in remission at the time of infusion impressively remained in remission longer than a comparator cohort.29 However, no survival advantage was observed in those who received T cells with active leukemia.28 In the current trial, mLSTs targeted multiple leukemia antigens and epitopes, thereby permitting inclusion of all individuals irrespective of HLA type. The infused mLSTs produced responses in select patients with active disease. By targeting naturally presented epitopes, we were also able to access “leukemia-selective” antigens that have minimal normal HSC expression in contrast with CAR-targets. With safety established, immediately available strategies can be combined (eg, epigenetic modifiers and salvage chemotherapy) to potentially improve the efficacy of mLSTs.
In 5 patients who relapsed after receiving mLSTs as an adjuvant therapy and in contrast to a matched cohort of consecutive patients treated after HCT/DLI, we found evidence of tumor evolution as a countermeasure to T-cell attack. Observed signatures included upregulation of PD-L1 and decrease/loss of major histocompatibility complex class 2 and/or target antigen expression on malignant cells, as well as disease relapse in immune-privileged sites, such as the CNS and bone.13-18 Other mechanisms may also have been responsible for relapses, and it should be noted that our findings are constrained by small samples and heterogeneity in patient profiles. These escape phenomena can be addressed with the application of clinically available agents including checkpoint inhibitors,30,31 IFNs (upregulate HLA expression),32 and epigenetic therapies to alter TAA expression,33-37 combined with higher doses of mLSTs. Indeed, DLIs and epigenetic modifiers have been previously combined. For example, in 2 trials, administration of 5-azacytidine36 or panabinostat37 with DLIs demonstrated lower than expected relapse rates of 27% and 20%, respectively. In the current study, we reenrolled and successfully re-treated 3 of those patients with mLSTs (1 at the same dose level and 2 others at higher dose levels). In all 3 cases before mLST infusion we addressed potential mechanisms of failure: intrathecal chemotherapy in patient 6 with CNS-only disease, withdrawal of corticosteroids in patient A1, and higher doses of cells in patient 9. In addition, our platform can support the generation of mLSTs targeting a broader spectrum of TAAs, such as the MAGE family of antigens,35 PR1,38 and/or cyclin A1,39,40 especially given that inter- and intrapatient variability in the expression of individual leukemic blasts can be expected.39-41
In summary, administration of mLSTs at doses comparable to those in DLIs proved safe and produced direct anticancer effects in chemoresistant AML/MDS relapses after HCT. These results will be subjected to validation in randomized, multicenter trials, which are currently under way. Nevertheless, this study provides compelling evidence that mLSTs can be safely administered as a single agent or in combination to effectively produce and/or sustain long-term remissions, irrespective of a patient’s HLA type. Given the simplicity and robustness of mLST manufacture (without gene modification) that can be made available to all patients with AML/MDS who undergo HCT, as well as the demonstrated antileukemia effects, we believe that mLSTs addresses a major unmet need in the management of AML/MDS after HCT.
The clinical protocol, individual level patient data, and correlation analysis are provided in the publication, with raw data available for all. Reasonable requests for additional raw/trial data will be provided upon e-mail inquiry (lulla@bcm.edu).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Walter Mejia for assistance with preparation of the figures.
This work was supported by Leukemia and Lymphoma Society Specialized Center of Research (SCOR) awards (H.E.H.), the Leukemia Lymphoma Society/Rising Tide Foundation (A.M.L.), an ASH Scholar Award (P.D.L.), a Leukemia Texas Research grant (P.D.L.), an American Society of Blood and Marrow Transplantation (ASBMT) New Investigator Award (P.D.L.), an Edward P. Evans Foundations Discovery Research Grant (P.D.L.), the Caroline Wiess Law Fund for Research in Molecular Medicine and the L. E. and Josephine S. Gordy Memorial Cancer Research Fund (P.D.L.), the Cancer Prevention and Research Institute of Texas (CPRIT) Texas Access to Cancer Cell Therapies (TACCT) (A.P.G.), a CPRIT Early Career Clinical Investigator Award (grant RP200584 to P.D.L.), the Frank Stahl gift to Houston Methodist Hospital for leukemia research, and the Dan L. Duncan Comprehensive Cancer Center for application of the shared resources from a support grant from the National Institutes of Health, National Cancer Institute (P30CA125123).
Authorship
Contribution: A.M.L., H.E.H., M.K.B., P.D.L., S.N., A.P.G., and S.G. conceived and designed the study; B.G. and C.R. provided administrative and regulatory support; P.D.L., S.N., R. Kamble, S.G., G.C., C.A.R., R. Krance, L.H., and J.R. recruited and cared for the patients; all authors collected and assembled the data; P.D.L., A.M.L., H.E.H., M.K.B., S.V., I.T., S.L., M.K., A.W., J.F.V., T.W., B.C., and M.W. analyzed and interpreted the data; P.D.L., A.M.L., and S.V. wrote the manuscript; and all authors approved the final draft of the manuscript and are accountable for all aspects of the work.
Conflict-of-interest disclosure: Baylor College of Medicine has a financial relationship with Marker Therapeutics. B.G. is a consultant for Marker Therapeutics. A.M.L., J.F.V., M.K.B., and H.E.H. are cofounders and equity holders in AlloVir Inc and Marker Therapeutics. J.F.V. is an employee of Marker Therapeutics, which aspires to commercialize the described approach. M.K.B. also serves on the advisory board for Tessa Therapeutics, Allogen and Unum. H.E.H. has served on advisory boards for Gilead Biosciences, Novartis, PACT Pharma, Mesoblast, Kiadis, and Tessa Therapeutics and has received research support from Tessa Therapeutics and Kuur Therapeutics. S.G. has patent applications in the fields of T-cell and/or gene therapy for cancer; he has a research collaboration with Tessa Therapeutics, is a DSMB member of Immatics, and serves on the scientific advisory board of Tidal. The remaining authors declare no competing financial interests.
The current affiliation for S.G. is Department of Bone Marrow Transplant and Cellular Therapy, St Jude Children’s Research Hospital, Memphis, TN.
The current affiliation for B.C. is Ochsner Health System, New Orleans, LA.
Correspondence: Premal D. Lulla, Center for Cell and Gene Therapy, Feigin Center FC1780.06, 1102 Bates Ave, Houston, TX 77030; e-mail: lulla@bcm.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal