

TO THE EDITOR:
Nucleophosmin (NPM1), encoding for a primarily nucleolar localized multifunctional protein,1 is the most commonly mutated gene (∼30% of cases) in adult acute myeloid leukemia (AML). The mutations (NPM1c) result in their aberrant cytoplasmic localization.2,3 Interestingly, the interaction of mixed-lineage leukemia (MLL1) with menin in NPM1c AML shares a common HOX gene signature and dependencies with that of MLL rearrangements (MLL1-r) with menin.4 Indeed, inhibition of menin demonstrated antileukemia activity in NPM1c and MLL-r AML.5-12 NPM1 mutations in AML frequently occur in patients carrying other mutations, such as FLT3.3 NPM1c cooperates with FLT3-internal tandem duplication (ITD), as well as the tyrosine kinase domain (TKD) mutations, to promote leukemogenesis.13-15 Coinhibition of menin and FLT3 demonstrated enhanced antileukemia activity in MLL-r/FLT3–mutated and NPM1c/FLT3-mutated AML.16,17
Targeting Bcl-2, a critical AML survival factor, has emerged as a promising therapeutic option for patients with the disease. However, despite a major increase in complete remission/complete remission with incomplete hematologic recovery rates by combining the Bcl-2 inhibitor venetoclax with a hypomethylating agent, most patients develop resistance and, ultimately, relapse.18-20 Nevertheless, venetoclax has become a mainstay for combinatorial AML therapies. We investigated the antileukemic activity, potential synergism, and mechanisms of the combination of the menin-MLL1 inhibitor SNDX-50469, which is an equipotent surrogate of the clinical compound SNDX-5613, and venetoclax in vivo in an NPM1c/FLT3-ITD/TKD patient-derived xenograft (PDX) model and in vitro in primary NPM1c/FLT3–mutated AML patient samples.
Mouse experiments were performed following MD Anderson’s Institutional Animal Care and Use Committee–approved protocols. The PDX (DFAM-16835) was obtained from the PRoXe (www.proxe.org) depository.21 Engrafted NSG mice were treated with 0.05% or 0.1% SNDX-50469–spiked chow, venetoclax (50 mg/kg, daily, oval gavage), or 0.1% SNDX-50469 plus venetoclax (Figure 1A). At 2 weeks, SNDX-50469 (0.05% or 0.1%; P < .0001) or venetoclax (P = .0012) significantly decreased circulating blasts as assessed by flow cytometric measurement of human CD45+ (huCD45+) cells. The higher dose was more effective (P = .05), and the combination was significantly more effective (P < .0001) than 0.1% SNDX-50469 or venetoclax (Figure 1B). At 4 weeks, SNDX-50469, alone or in combination with venetoclax, significantly (P < .0001) diminished circulating leukemia cells, whereas venetoclax alone was ineffective (Figure 1C).
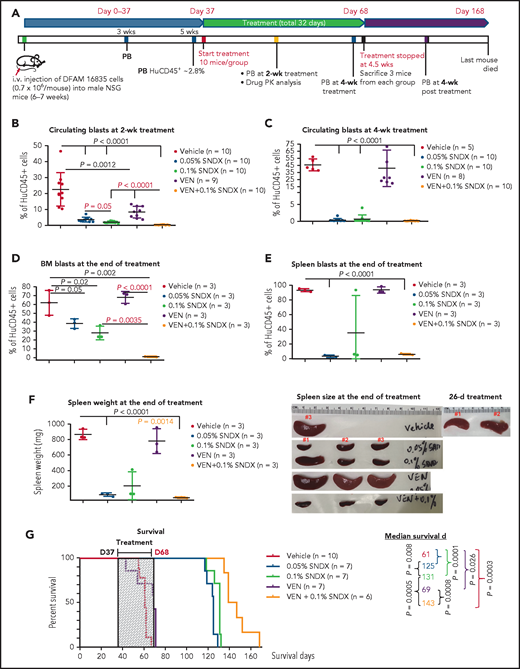
Menin inhibition demonstrates antileukemia activities and prolongs mouse survival, which is further enhanced by Bcl-2 inhibition in an NPM1c/FLT3-ITD/TKD PDX model. (A) The mouse model and experimental scheme. Percentage of huCD45+ cells in peripheral blood at 2 weeks (B) and at 4 weeks (C) and at the end of the treatment in BM (D) and spleen (E), as determined by flow cytometry. (F) Spleen weight and size at the end of the treatment. (G) Survival curve. Mouse survival was estimated using the Kaplan-Meier method, and survival data were analyzed using the log-rank test. Differences between groups were determined using the Student t test. Values of P ≤.05 were considered statistically significant. D, day; PB, peripheral blood; PK, pharmacokinetics; SNDX, SNDX-50469; VEN, venetoclax.
Menin inhibition demonstrates antileukemia activities and prolongs mouse survival, which is further enhanced by Bcl-2 inhibition in an NPM1c/FLT3-ITD/TKD PDX model. (A) The mouse model and experimental scheme. Percentage of huCD45+ cells in peripheral blood at 2 weeks (B) and at 4 weeks (C) and at the end of the treatment in BM (D) and spleen (E), as determined by flow cytometry. (F) Spleen weight and size at the end of the treatment. (G) Survival curve. Mouse survival was estimated using the Kaplan-Meier method, and survival data were analyzed using the log-rank test. Differences between groups were determined using the Student t test. Values of P ≤.05 were considered statistically significant. D, day; PB, peripheral blood; PK, pharmacokinetics; SNDX, SNDX-50469; VEN, venetoclax.
Flow cytometric analysis revealed that, at the end of the treatment, SNDX-50469 at 0.05% (P = .05) or 0.1% (P = .02) partially decreased bone marrow (BM) leukemia cells. Although the higher dose tended to be more effective, no statistical significance was reached. Venetoclax alone did not show any activity, but it markedly diminished BM leukemia burden when combined with 0.1% SNDX-50469 (P = .0035 vs 0.1% SNDX-50469) (Figure 1D). Venetoclax alone also lacked activity in the spleen, whereas SNDX-50469, alone or in combination with venetoclax, largely reduced splenic huCD45+ cells (Figure 1E), as well as spleen weight and size (Figure 1F). Of note, 1 mouse treated with 0.1% SNDX-50469 exhibited high blasts and an enlarged spleen; it also had relatively higher BM huCD45+ cells (Figure 1D), which were consistent with hematoxylin and eosin staining (supplemental Figure 1, available on the Blood Web site).
SNDX-50469 (0.05% or 0.1%) significantly extended mouse survival (median, 125 days or 131 days, respectively, vs 61 days for controls; both P = .0001), and the higher dose showed increased benefit (P = .008). Venetoclax alone minimally prolonged survival (median, 69 days, P = .026) vs controls. However, mice treated with 0.1% SNDX-50469 plus venetoclax experienced a more than twofold increase in their overall survival (median, 143 days) compared with untreated mice (P = .0003) or venetoclax-treated mice (P = .0008), which was longer than that of mice treated with 0.1% SNDX-50469 alone (P = .0005).
At the end of the therapy, the treatment effects on leukemia blasts and phenotypically defined leukemia stem/progenitor cells, along with protein expression of BM leukemia cells, were assessed by cytometry by time of flight analysis, as described in detail previously.22 The cytometry by time of flight panel is shown in supplemental Table 1. Analysis of huCD45+ cells showed that SNDX-50469 altered the cellular composition and that venetoclax had only a minimal effect on leukemia cells, whereas the combination effectively eliminated the leukemia cells (Figure 2A). PhenoGraph clustering, based on cell surface marker expression, grouped huCD45+ cells into CD34+CD38+, CD34+CD38+CD123+, CD34+CD38+CD123+Tim3+, CD34+CD38−, CD34+CD38−CD123+, and CD34+CD38−CD123+Tim3+ populations (Figure 2B). SNDX-50469 at 0.05%, and more so at 0.1%, partially suppressed bulk leukemia cells and effectively targeted CD34+ CD38+/CD34+CD38+CD123+/CD34+CD38+CD123+Tim3 cells. At 0.1%, SNDX-50469 was able to reduce CD34+CD38−/CD34+CD38−CD123+ cells but not CD34+CD38−CD123+Tim3+ cells. Venetoclax had no activity against bulk leukemia and only partial activity against CD34+CD38+/CD34+CD38+CD123+/CD34+CD38+CD123+Tim3 cells. However, it was active against the CD34+CD38−/CD34+CD38−CD123+/CD34+CD38−CD123+Tim3+ population. The combination of SNDX-50469 (0.1%) and venetoclax was most effective in eliminating all cell types, including leukemia stem/progenitor cells (Figure 2C). Protein analysis of leukemia cells (Figure 2D) demonstrated that SNDX-50469, and even more so the combination, decreased Bcl-2 and Bcl-xL and increased Bim. Furthermore, the combination decreased Bcl-2A1, a resistance factor for Bcl-2 inhibition.23,24 Protein analysis of CD34+CD38+ and CD34+CD38− cells (supplemental Figure 2) revealed that SNDX-50469 increased multiple proapoptotic proteins. SNDX-50469 decreased Bcl-2 in CD34+CD38+ cells, but not markedly so in CD34+CD38− cells, which may explain, in part, its effectiveness in CD34+CD38+ cells rather than in the CD34+CD38− population. Because CD34+CD38+ and CD34+CD38− cell numbers were extremely low in the combination-treated group, protein levels in these cells may be difficult to correlate with the observed outcomes.

Menin and Bcl-2 inhibition targets leukemia and stem/progenitor cells, modulates Bcl-2 protein levels in BM cells, and exhibits enhanced antileukemia activity in primary AML patient samples. Cell populations were PhenoGraph clustered based on cell surface markers. Cisplatin-low viable single cells were gated with FlowJo software v10.7 and exported as flow cytometry standard data for subsequent analysis in Cytofkit.25 Cell populations identified and embedded by PhenoGraph in the “Cytofkit_analyzedFCS” files were gated in FlowJo to quantify marker expression. ArcSinh-transformed counts for each protein expression in desired cell populations were visualized using heat maps. (A) huCD45+ cells in various treatment groups. (B) Clusters of leukemia cells and leukemia stem/progenitor cells. (C) Percentage of viable leukemia cells and leukemia stem/progenitor cells in each treatment group. (D) Protein expression in huCD45+ cells in various treatment groups. (E) Percentage of huCD11b+CD45+ cells in each treatment group. (F) Mononuclear cells from NPM1/FLT3-mutant AML patient samples (n = 5) were treated with SNDX-50469, venetoclax, or both for 24 hours. Apoptosis, cell viability, and p-FLT3 levels were determined by flow cytometry. Samples were obtained after acquiring written informed consent following the MD Anderson Cancer Center Institutional Review Board–approved protocol and in accordance with the Declaration of Helsinki. Differences between groups were determined using the Student t test. Values of P ≤.05 were considered statistically significant. con/CON, control; SNDX, SNDX-50469; VEN, venetoclax.
Menin and Bcl-2 inhibition targets leukemia and stem/progenitor cells, modulates Bcl-2 protein levels in BM cells, and exhibits enhanced antileukemia activity in primary AML patient samples. Cell populations were PhenoGraph clustered based on cell surface markers. Cisplatin-low viable single cells were gated with FlowJo software v10.7 and exported as flow cytometry standard data for subsequent analysis in Cytofkit.25 Cell populations identified and embedded by PhenoGraph in the “Cytofkit_analyzedFCS” files were gated in FlowJo to quantify marker expression. ArcSinh-transformed counts for each protein expression in desired cell populations were visualized using heat maps. (A) huCD45+ cells in various treatment groups. (B) Clusters of leukemia cells and leukemia stem/progenitor cells. (C) Percentage of viable leukemia cells and leukemia stem/progenitor cells in each treatment group. (D) Protein expression in huCD45+ cells in various treatment groups. (E) Percentage of huCD11b+CD45+ cells in each treatment group. (F) Mononuclear cells from NPM1/FLT3-mutant AML patient samples (n = 5) were treated with SNDX-50469, venetoclax, or both for 24 hours. Apoptosis, cell viability, and p-FLT3 levels were determined by flow cytometry. Samples were obtained after acquiring written informed consent following the MD Anderson Cancer Center Institutional Review Board–approved protocol and in accordance with the Declaration of Helsinki. Differences between groups were determined using the Student t test. Values of P ≤.05 were considered statistically significant. con/CON, control; SNDX, SNDX-50469; VEN, venetoclax.
At the end of treatment, although the combination tended to be more effective than was SNDX-50469 alone, it did not reach statistical significance with regard to suppressing circulating and spleen blasts or decreasing spleen weight or size. This is likely due, in part, to the effectiveness of menin inhibition alone (Figure 1C,E-F). However, the combination markedly reduced circulating blasts at an earlier time, more effectively targeting BM leukemia blasts and stem/progenitor cells, and it modulated cellular protein levels compared with SNDX-50469 alone. This translated to considerably longer overall mouse survival (Figures 1B,D,G and 2).
Contrary to reports that menin inhibition in NPM1c/FLT3-mutated AML targets FLT3,16,17 we observed that phosphorylated (p)-FLT3 increased in SNDX-50469–treated cells, especially in the combination group, which likely caused increases in Mcl-1 and contributed to subsequent resistance. Decreased FLT3 expression was observed in vitro in cell lines following short-term treatment with menin inhibitor,16,17 whereas our results were obtained in vivo from mice treated for 1 month and reflect the single-cell proteomics of the surviving cells. The increase in p-FLT3 could be induced by BM environmental factors, or it could be a resistance mechanism in the surviving cells. Higher levels of p-FAK and CD44 may indicate stromal interactions activated to enhance survival. Furthermore, we observed increased huCD11b levels (Figure 2D) and huCD11b+ populations in SNDX-50469–treated mouse BM cells (Figure 2E), which is consistent with increased myeloid differentiation reported in menin inhibitor–treated NPM1c AML.12
To ensure proper drug intake, blood samples were taken from mice fed SNDX-50469–spiked chow, and the drug level was determined in the plasma (n = 5). Dose-dependent plasma levels of SNDX-50469 were observed, which were not affected by treatment with venetoclax (supplemental Figure 3). The combination treatment caused weight loss, which could result in an underestimation of the combinatorial treatment efficacy, and decreased mouse CD45+ cells. However, both metrics recovered over time (supplemental Figures 4 and 5).
To further demonstrate the enhanced antileukemia activity of combined menin and Bcl-2 inhibition, we treated NPM1c/FLT3-mutated primary AML patient samples (supplemental Table 2) with SNDX-50469, venetoclax, or both. The combination of SNDX-50469 (62.5 nM) and venetoclax (2.5 nM) was dramatically more effective in increasing apoptosis and decreasing cell viability than was either agent alone, and the efficacy was enhanced with higher doses of each agent (ie, SNDX-50469 [250 nM] plus venetoclax [10 nM]) (Figure 2F). SNDX-50469 (62.5 nM) decreased p-FLT3 levels in 3 of 5 samples. However, when the 2 agents were combined, a statistically significant (P = .0027) reduction in p-FLT3 was observed in all samples (Figure 2F). These results were comparable to published data;16,17 however, as mentioned, they were obtained using short-term in vitro treatments.
Collectively, we demonstrate that menin inhibition exhibits strong antileukemia activity and significantly prolonged mouse survival, which was further enhanced when combined with venetoclax in an NPM1c/FLT3-ITD/TKD AML PDX model. Menin inhibition preferentially targeted CD34+CD38+ cells, whereas venetoclax targeted CD34+CD38− cells. Only the combination effectively eliminated bulk and CD34+CD38+/CD34+CD38− stem/progenitor cells. Mechanistically, menin inhibition decreased multiple antiapoptotic Bcl-2 proteins and concomitantly increased proapoptotic Bcl-2 proteins that seemingly enhanced the activity of venetoclax. It is not known whether extended treatment would further enhance the survival benefit of this combination. Additionally, the combination was more effective in vitro in primary NPM1c/FLT3 mutant AML cells. Our study further validates menin as a therapeutic target and demonstrates that its inhibition synergizes with venetoclax in NPM1/FLT3-mutated AML, which warrants clinical evaluation. Given the high activity of p-FLT3 at the end of in vivo treatment and the reported synergism of menin and FLT3 inhibition, a triple-drug combination may further enhance the activity of menin inhibition in FLT3-mutant AML. It is important to point out that these results were obtained in 1 NPM1c/FLT3-ITD/TKD PDX model and in vitro with 5 primary patient samples (4 NPM1c/FLT3-ITD/TKD samples and 1 NPM1c/FLT3-ITD sample). Further studies in additional models are warranted to confirm these findings.
Acknowledgments
The authors thank Numsen Hail for assistance with editing the manuscript.
This work was supported, in part by research funding from Syndax (B.Z.C.), a National Institutes of Health, National Cancer Institute Cancer Center Support Grant (P30CA016672), and the Paul and Mary Haas Chair in Genetics (M.A.).
Authorship
Contribution: B.Z.C. conceptualized the study, analyzed data, and wrote the manuscript; W.T. and P.Y.M. performed experiments and analyzed data; L.B.O. and D.M. performed experiments; G.M. and P.O. discussed the study concept, provided materials, and edited the manuscript; and M.A. conceptualized the study, interpreted data, and edited the manuscript.
Conflict-of-interest disclosure: B.Z.C. received research funding from Syndax. G.M. and P.O. are employees of Syndax. M.A. is consultant for Syndax. The remaining authors declare no competing financial interests.
Correspondence: Bing Z. Carter, Section of Molecular Hematology and Therapy, Department of Leukemia, Unit 448, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: bicarter@mdanderson.org; and Michael Andreeff, Section of Molecular Hematology and Therapy, Department of Leukemia, Unit 448, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Houston, TX 77030; e-mail: mandreef@mdanderson.org.
Data sharing requests should be sent to Michael Andreeff (mandreef@mdanderson.org).
The online version of this article contains a data supplement.
