

Key Points
Somatic uniparental disomy in the bone marrow generated an EFL1 allele combination that confers selective advantages.
Nevertheless, the resulting homozygous EFL1 allele in the hematopoietic system is hypomorphic and still responsible for SDS features.

Abstract
Shwachman-Diamond syndrome (SDS; OMIM #260400) is caused by variants in SBDS (Shwachman-Bodian-Diamond syndrome gene), which encodes a protein that plays an important role in ribosome assembly. Recent reports suggest that recessive variants in EFL1 are also responsible for SDS. However, the precise genetic mechanism that leads to EFL1-induced SDS remains incompletely understood. Here we present 3 unrelated Korean SDS patients who carry biallelic pathogenic variants in EFL1 with biased allele frequencies, resulting from a bone marrow–specific somatic uniparental disomy in chromosome 15. The recombination events generated cells that were homozygous for the relatively milder variant, allowing for the evasion of catastrophic physiologic consequences. However, the milder EFL1 variant was still solely able to impair 80S ribosome assembly and induce SDS features in cell line and animal models. The loss of EFL1 resulted in a pronounced inhibition of terminal oligopyrimidine element–containing ribosomal protein transcript 80S assembly. Therefore, we propose a more accurate pathogenesis mechanism of EFL1 dysfunction that eventually leads to aberrant translational control and ribosomopathy.
Introduction
Patients clinically diagnosed with Shwachman-Diamond syndrome (SDS; OMIM #260400) present with a constellation of disorders, such as hematologic manifestations, exocrine pancreatic dysfunction with fatty infiltration, and skeletal dysplasia that results in short stature.1-3 The hematologic manifestations include neutropenia or, less severely, thrombocytopenia and anemia, with a predisposition for myelodysplastic syndrome and acute myeloid leukemia transformations.4,5 Variants in SBDS (Shwachman-Bodian-Diamond syndrome gene), which encodes a protein that plays an important role in ribosome assembly, are mainly responsible for the disease.1,6-8 Thus, SDS is considered to be a ribosomopathy, which is a collective term that is used to describe a group of congenital disorders caused by problems in ribosome biogenesis, assembly, or function.9 Moreover, ∼10% of clinically diagnosed SDS cases do not contain any pathogenic SBDS variants, suggesting the existence of additional genetic mechanisms that lead to the disorder.1,6 Recent reports demonstrate that variants in genes other than SBDS, namely EFL1, DNAJC21, and SRP54, are implicated in bone marrow failure syndrome and SDS.10-15 Homozygous variants of EFL1 cause an SDS-like syndrome in a recessive manner, which is highlighted by the observation that 5 of the 7 reported kindreds harbor homozygous EFL1 variants (supplemental Table 1).10,11,15 EFL1 directly interacts with SBDS to release eukaryotic translation initiation factor 6 (eIF6) from the 60S ribosomal subunit for 80S ribosomal assembly.7,16 Therefore, whether there is an additional genetic mechanism that leads to SDS in outbred populations other than through homozygous pathogenic variants in EFL1 must be further investigated.
As more human genomes with or without clinical significances have continued to be sequenced, it has become clear that variants of unknown significance pose a substantial obstacle in the interpretation of genotype-phenotype relationships. Because many variants are believed to possess the ability to cause alterations at the molecular level but only with subclinical levels of severity, numerous scenarios that enable variants of unknown significance to acquire clinical significance have been postulated. One of these scenarios involves the assessment of somatically acquired uniparental disomy (UPD) in the hematopoietic system, although only a small number of instances have been previously reported. Notable examples include myeloid neoplasia,17,18 immunodeficiency,19 and a single case of sickle cell disease.20
In this study, we demonstrate a disease-causing mechanism in patients who inherited compound heterozygous (CH) variants in EFL1. A mosaic UPD caused a loss of heterozygosity (LOH) in the EFL1 locus in the bone marrow and blood, simultaneously homozygosing the less damaging variant and decreasing the representation of the more damaging variant to avoid worse hematologic phenotypes. However, this still led to EFL1 dysfunction in the bone marrow and resulted in SDS features. We further demonstrate that the remaining variant caused by the UPD was a hypomorph and pathogenic, by investigating the molecular mechanism of the EFL1 dysfunction in cell and animal models. Therefore, this study provides a template for searching for a pathologic variant that is caused by a nonconventional pathway. Using this method may increase the probability of both identifying the genetic cause and improving our understanding of the SDS disease mechanism. This approach could benefit additional patients with severe hematologic abnormalities.
Methods
Patient recruitment and sampling
Patient enrollment and sampling were conducted under the approval of the Institutional Review Board of Seoul National University Hospital (H-1408-014-599). Patients or their parents provided informed consent and signed for genetic testing and collection of available tissues. Biopsy samples of I-1 (duodenum and colon) were retrieved from the Department of Pathology of Seoul National University for research purposes.
WES and variant calling
Trio whole-exome sequencing (WES) was performed on 1 kindred (I-1, I-2, and I-3), and singleton WES was performed for the others (II-1 and III-1) at Theragen Etex (Suwon, Korea) using genomic DNA extracted from whole blood. Exome was captured using the SeqCap EZ Exome v2 Kit (Roche Sequencing, Madison, WI) or SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA) and sequenced using a HiSeq 2500 or HiSeq 4000 (Illumina, Inc, San Diego, CA). Paired-end sequencing was performed with read lengths of 75 or 100 base pairs. The raw reads were aligned by BWA-MEM.21 Variants were called by GATK HaplotypeCaller (version 3.8) and annotated by in-house pipeline and SnpEff.21-23
Polysome profiling
To maintain the binding of messenger RNA (mRNA) to ribosome subunits, cycloheximide (100 μg/mL; Sigma-Aldrich, St Louis, MO) was added to cell culture media and incubated for 10 minutes at 37°C. After incubation, cells were washed with cold phosphate-buffered saline including cycloheximide (10 μg/mL) twice and then lysed with 1 mL of polysome lysis buffer (20 mM of HEPES [pH 7.6], 5 mM of magnesium chloride, 125 mM of potassium chloride, 1% NP-40, and 2 mM of DTT) supplemented with cycloheximide (100 μg/mL), protease inhibitor cocktail (EDTA free; Roche), and RNase inhibitor (Invitrogen, Carlsbad, CA), on ice. Cell lysates were tumbled for 20 minutes at 4°C and centrifuged at 13 200 rpm for 20 minutes. The supernatants were fractionated in 17.5% to 50.0% linear sucrose gradients by ultracentrifugation (35 000 rpm for 160 minutes) in a Beckman ultracentrifuge using an SW41-Ti rotor. Gradients were eluted with a gradient fractionator (Brandel, Gaithersburg, MD) and monitored with a UA-5 detector (ISCO). Equal volume of each polysome fraction was used for determining the level of eIF6 by western blot analysis.
Mouse strain construction, maintenance, and experiments
All mouse experiments were performed under the standard protocols approved by the Institutional Animal Care and Use Committee (17-0148-S1A0). The Efl1 knockout (Efl1−) strain was constructed by introducing a 10-base deletion in the 10th exon of the gene in the C57BL6/J strain using the CRISPR/Cas9 system in Macrogen (Seoul, South Korea). One-cell embryos were microinjected with 2 single guide RNAs (5'-ACTTCTTTAGGATTAAAAATTGG-3' and 5'-CCGAGGACAGCGTGGGATATGGG-3') and Cas9 protein mixture, incubated, and transplanted into pseudopregnant recipient ICR mice. The Efl1 knock-in (Efl1p.Thr1076Ala) variant was generated in C57BL6/J mice at the University of Utah Mutation Generation and Analysis Core using 2 single guide RNAs (5'-GTTCTGGGTGCCGACCACGG-3' and 5'-GTGCAGGTACTCCTCCTCCG-3') and in the presence of oligodeoxynucleotides, including the ACC>GCC change mimicking the human EFL1p.Thr1069Ala and an MboI site for genotyping. Genotyping strategies of the wild-type and mutant alleles and phenotypic evaluations are described in the supplemental Data, available on the Blood Web site.
Results
SDS patients without SBDS variants
We recruited 3 unrelated and nonconsanguineous Korean SDS patients without plausible recessive mutations in SBDS (Figure 1A; supplemental Table 2; supplemental Figure 1; data supplement). Proband I-1 was a 3-year-old boy who had severe intrauterine growth retardation that resulted in a preterm delivery (35 + 3 weeks) and a birth weight of 1.7 kg. He had thrombocytopenia, neutropenia, and anemia at 2 months of age. A bone marrow examination performed at 6 months of age revealed hypocellularity, reduced megakaryocytes, and an increase in iron storage. He also had pancreatic lipomatosis, along with an exocrine pancreatic insufficiency and metaphyseal chondrodysplasia (Figure 1A; supplemental Figure 2). Proband II-1 was a 9-year-old girl who had severe intrauterine growth retardation that resulted in a preterm delivery (36 weeks) and a birth weight of 1.6 kg. She had diffuse fatty infiltration of the pancreas and metaphyseal chondrodysplasia that was accompanied by osteopenia and short stature (Figure 1A). Her sister was unaffected. Proband III-1 was a 25-year-old man who did not have any perinatal problems except for a low birth weight (40 + 4 weeks; 2.4 kg). At 2 years of age, he had pancreatic exocrine and endocrine insufficiencies, thrombocytopenia, anemia, intermittent neutropenia, metaphyseal chondrodysplasia, and ichthyosis. Later, he developed osteoporosis, hepatomegaly, and a total fatty change of the pancreas (Figure 1A).
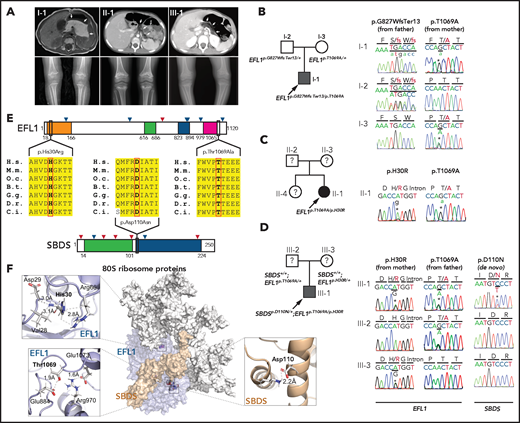
EFL1 variants in SBDS− SDS patients. (A) Noncontrast T1-weighted abdominal magnetic resonance (I-1) or computed tomography images (II-1 and III-1) showing a diffuse enlargement with lipomatosis of the pancreas (arrows; upper) and both knees with metaphyseal widening and irregularities in the femora in the 3 patients and associated genu varum in III-1 (lower). (B-D) Pedigrees and Sanger sequencing traces showing the inherited EFL1 variants and the de novo SBDSp.Asp110Asn variant in the 3 families. DNA was extracted from whole-blood samples. Note that the lower-case nucleotide letters were used on top of the Sanger traces to reflect their minor allelic representation in the patient samples. (E) The residues with nonsynonymous changes in EFL1 and SBDS are evolutionarily conserved. The arrowheads on the protein maps denote previously reported pathogenic variants (blue, missense; red, loss of function). (F) Molecular modeling–based structural analysis using PDB 5ANC.7 The insets show the detailed interactions involving EFL1 His30, Thr1069, and SBDS Asp110. B.t., Bos taurus; C.i., Ciona intestinalis; D.r., Danio rerio; G.g., Gallus gallus; H.s., Homo sapiens; M.m., Mus musculus; O.c., Oryctolagus cuniculus.
EFL1 variants in SBDS− SDS patients. (A) Noncontrast T1-weighted abdominal magnetic resonance (I-1) or computed tomography images (II-1 and III-1) showing a diffuse enlargement with lipomatosis of the pancreas (arrows; upper) and both knees with metaphyseal widening and irregularities in the femora in the 3 patients and associated genu varum in III-1 (lower). (B-D) Pedigrees and Sanger sequencing traces showing the inherited EFL1 variants and the de novo SBDSp.Asp110Asn variant in the 3 families. DNA was extracted from whole-blood samples. Note that the lower-case nucleotide letters were used on top of the Sanger traces to reflect their minor allelic representation in the patient samples. (E) The residues with nonsynonymous changes in EFL1 and SBDS are evolutionarily conserved. The arrowheads on the protein maps denote previously reported pathogenic variants (blue, missense; red, loss of function). (F) Molecular modeling–based structural analysis using PDB 5ANC.7 The insets show the detailed interactions involving EFL1 His30, Thr1069, and SBDS Asp110. B.t., Bos taurus; C.i., Ciona intestinalis; D.r., Danio rerio; G.g., Gallus gallus; H.s., Homo sapiens; M.m., Mus musculus; O.c., Oryctolagus cuniculus.
Identification of mosaic EFL1 variants
To identify the genetic factors that predisposed the 3 patients to SDS, we exome sequenced the patients, and the available parental DNA was extracted from whole blood (supplemental Table 3). Notably, the heterozygous p.Thr1069Ala variant of EFL1 (chr15:82 422 872 T>C, hg19, NM_024580.5:c.3205A>G) was identified in all 3 patients (Figure 1B-D, Table 1; supplemental Table 4), which was not previously found in patients with SDS. Based on gnomAD, this is a low-frequency variant that was carried by 3 individuals among 17 972 alleles in the East Asian (EA) population (EA AF, 1.7 × 10−4).24 Under the assumption that EFL1 variants function in a recessive manner, we sought to ascertain additional variants that may pose increased damage to the gene function. Remarkably, we found second variants in EFL1 in all 3 patients. These were not initially detected, either because of a low number of variant-supporting reads in the proband or because of low mapping quality caused by the sequence similarity between the EFL1 and EFL1P1 loci. Proband I-1 carried paternally originating frameshift variant p.Gly827TrpfsTer13 (chr15:82 444 316 C>CA, hg19, NM_024580.5:c.2478dupT; gnomAD EA AF, 0) with a minor AF of 8.3%, and probands II-1 and III-1 carried inherited missense variant p.His30Arg (chr15: 82 554 031 T>C, hg19, NM_024580.5:c.89A>G; gnomAD EA AF, 5.1 × 10−5) with minor AFs of 14.8% and 36.8%, respectively (Table 1). In addition, proband III-1 harbored a de novo heterozygous SBDS p.Asn110Asp variant (Figure 1D; supplemental Figure 3), which had never been seen in the control databases. We then noted that the nonreference allele of EFL1 p.Thr1069Ala for I-1 and II-1 was dominantly covered compared with the reference allele (Table 1), which caused it to be an incomplete homozygous variant, whereas the ratio was comparable in III-1 (Figure 1B-D; Table 1). All the EFL1 variants were equally represented in the parental carriers. The patients did not carry any variant in other SDS-associated genes, including DNAJC21 and SRP54 (data not shown). The 2 EFL1 amino acid residues harboring the missense variants (His30 and Thr1069) are highly conserved throughout evolution and are predicted to be pathogenic (Figure 1E; supplemental Table 5). The protein structure analysis suggested that the His30 and Thr1069 residues form hydrogen bonds with neighboring residues, which would presumably confer stability to the protein structure. Notably, previously reported pathogenic residues Cys883 and Arg970 lie close to Thr1069 (Figure 1F; supplemental Figure 4).15 The SBDS Asn110 reside is also conserved among the vertebrate species (Figure 1E; supplemental Table 5) and is expected to interact with neighboring amino acids, including Lys151 (Figure 1F).
Coverage depths and AFs of EFL1 variants
| Patient | Variant in EFL1 | Method | Allele coverage depths | AF in gnomAD | |||
|---|---|---|---|---|---|---|---|
| Patient | Mother | Father | Overall | EA | |||
| I-1 | p.Thr1069Ala (M) | WES | 10:64 (86.5) | 39:40 (50.1) | 33:0 (0) | 3.2 × 10−5 | 1.7 × 10−4 |
| Amplicon sequencing | 23 914:131 714 (84.6) | 63 582:63 954 (50.1) | 121 502:507 (0.4) | ||||
| p.Gly827 TrpfsTer13 (P) | WES | 22:2 (8.3) | 27:0 (0) | 9:14 (60.9) | 0 | 0 | |
| Amplicon sequencing | 243 610:43 042 (15.0) | 505 158:74 (0.01) | 221 532:218 976 (49.7) | ||||
| II-1 | p.Thr 1069Ala (U) | WES | 15:59 (79.7) | NA | NA | 3.2 × 10−5 | 1.7 × 10−4 |
| p.His30Arg (U) | WES | 127:22 (14.8) | NA | NA | 1.8 × 10−5 | 5.1 × 10−5 | |
| III-1 | p.Thr 1069Ala (P) | WES | 43:30 (41.1) | NA | NA | 3.2 × 10−5 | 1.7 × 10−4 |
| Amplicon sequencing | 135 196:149 160 (52.5) | 189 517:1786 (0.9) | 87 347:85 317 (49.4) | ||||
| p.His 30Arg (M) | WES | 74:43 (36.8) | N/A | NA | 1.8 × 10−5 | 5.1 × 10−5 | |
| Amplicon sequencing | 25 316:22 680 (47.3) | 119 382:121 508 (50.4) | 41 684:76 (0.2) | ||||
| Patient | Variant in EFL1 | Method | Allele coverage depths | AF in gnomAD | |||
|---|---|---|---|---|---|---|---|
| Patient | Mother | Father | Overall | EA | |||
| I-1 | p.Thr1069Ala (M) | WES | 10:64 (86.5) | 39:40 (50.1) | 33:0 (0) | 3.2 × 10−5 | 1.7 × 10−4 |
| Amplicon sequencing | 23 914:131 714 (84.6) | 63 582:63 954 (50.1) | 121 502:507 (0.4) | ||||
| p.Gly827 TrpfsTer13 (P) | WES | 22:2 (8.3) | 27:0 (0) | 9:14 (60.9) | 0 | 0 | |
| Amplicon sequencing | 243 610:43 042 (15.0) | 505 158:74 (0.01) | 221 532:218 976 (49.7) | ||||
| II-1 | p.Thr 1069Ala (U) | WES | 15:59 (79.7) | NA | NA | 3.2 × 10−5 | 1.7 × 10−4 |
| p.His30Arg (U) | WES | 127:22 (14.8) | NA | NA | 1.8 × 10−5 | 5.1 × 10−5 | |
| III-1 | p.Thr 1069Ala (P) | WES | 43:30 (41.1) | NA | NA | 3.2 × 10−5 | 1.7 × 10−4 |
| Amplicon sequencing | 135 196:149 160 (52.5) | 189 517:1786 (0.9) | 87 347:85 317 (49.4) | ||||
| p.His 30Arg (M) | WES | 74:43 (36.8) | N/A | NA | 1.8 × 10−5 | 5.1 × 10−5 | |
| Amplicon sequencing | 25 316:22 680 (47.3) | 119 382:121 508 (50.4) | 41 684:76 (0.2) | ||||
Data in “Allele coverage depths” column presented as ratio of reference allele/nonreference allele (% nonreference allele).
AF, allele frequency; M, maternal origin; NA, not available; P, paternal original; U, unknown origin.
To understand the genetic cause of this observation, we investigated whether large-scale structural variants exist that encompass the region. Indeed, all the patients carried a partial LOH in chromosome 15, where EFL1 resides (Figure 2A; supplemental Figures 5 and 6). This LOH was copy neutral and not seen in the healthy parents (supplemental Figure 7), suggesting that it was caused by a somatic UPD. This LOH of the EFL1 locus is not frequently found in healthy Korean individuals (1/3667 = 2.7 × 10−4; supplemental Figure 8). Also, according to a survey of hematopoietic chromosomal mosaicism events, 117 of 151 202 apparently healthy individuals carry a copy-neutral LOH or copy-number deletion of the EFL1 locus (7.7 × 10−4).25 Thus, the LOH of the EFL1 locus is a rare event. The sizes of the LOH intervals were variable among the patients (100%, 100%, and 27.8% of the entire chromosome span for I-1, II-1, and III-1, respectively; Figure 2A). We also sought to identify the haplotype origins of the variants that the probands carried and observed that the chromosomes that were dominantly represented (ie, the maternal chromosome for I-1 and the paternal chromosome for III-1) harbored EFL1p.Thr1069Ala, which was consistent with their higher coverage ratios compared with other variants as documented in the WES analysis (Figure 2A). To test if the UPD event occurred in a mosaic pattern and whether LOH-carrying and non–LOH-carrying cells coexist, a single-cell single-nucleotide polymorphism microarray experiment was performed using bone marrow (I-1) or cells from buccal swabs (III-1). As expected, complete LOHs in chromosome 15 were observed in a subset of the cells, confirming the mosaic UPD events that preferentially selected EFL1p.Thr1069Ala over the other variants (ie, EFL1p.Gly827TrpfsTer13 and EFL1p.His30Arg; Figure 2B-D; supplemental Figure 9). Therefore, cells containing the UPD of chromosome 15 are expected to be homozygous for the EFL1p.Thr1069Ala allele and be homozygous references for the other 2 alleles. II-1 was not tested, because a sample was unavailable. Next, we checked the spatial extent of the mosaic UPD by subjecting all available tissue samples from I-1 to a high-depth amplicon sequencing analysis (>10 000 × coverage depths). Variant AFs of p.Thr1069Ala in I-1 were ∼0.85 in the peripheral blood and bone marrow but ∼0.5 in most tissues. Conversely, AFs of p.Gly827TrpfsTer13 in the peripheral blood and bone marrow were ∼0.15 and displayed frequencies complementary to those of p.Thr1069Ala (Figure 2E). This observation suggested that the mosaic UPD was restricted at least to the bone marrow. These results were concordant with the Sanger sequencing results (supplemental Figures 10 and 11). The degree of mosaicism in the bone marrow tissue changed dynamically over the time course of I-1 but did not strongly correlate with the clinical status of the patient (supplemental Figure 12). These results suggest that CH variants that may disable EFL1 function and impair cell survival formed a cellular environment such that cells with (less damaging) recombinant alleles gained survival advantages over the parental ones (Figure 2F).
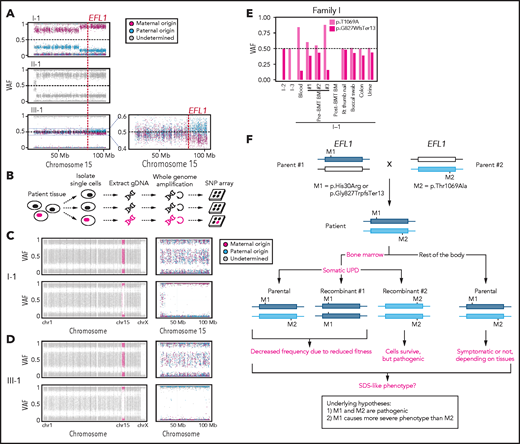
EFL1 variants display somatic mosaicism. (A) All 3 patients demonstrate a partial LOH in chromosome 15, as indicated by the deviation from variant AF (VAF) of 0.5. The variant origin is indicated by red or blue dots; parental samples were not available for II-1. The location of EFL1 is indicated by a red dotted line. (B) Schematic diagram of the single-cell LOH experiment shown in panels C and D, where a red cell symbolizes an LOH-carrying cell. (C-D) Single-cell LOH profiles from I-1 bone marrow (BM) (C) and III-1 buccal swab samples (D); the upper plots denote cells without LOH, and the lower plots represent cells with complete LOH in chromosome 15. The plots on the right show chromosome 15 with the variant origin shown in red or blue dots. (E) VAF of the EFL1 variant in multiple tissue samples from I-1. (F) Genetic process underlying selection of EFL1p.Thr1069Ala cells in the patients. BMT, bone marrow transplantation; gDNA, genomic DNA; SNP, single-nucleotide polymorphism.
EFL1 variants display somatic mosaicism. (A) All 3 patients demonstrate a partial LOH in chromosome 15, as indicated by the deviation from variant AF (VAF) of 0.5. The variant origin is indicated by red or blue dots; parental samples were not available for II-1. The location of EFL1 is indicated by a red dotted line. (B) Schematic diagram of the single-cell LOH experiment shown in panels C and D, where a red cell symbolizes an LOH-carrying cell. (C-D) Single-cell LOH profiles from I-1 bone marrow (BM) (C) and III-1 buccal swab samples (D); the upper plots denote cells without LOH, and the lower plots represent cells with complete LOH in chromosome 15. The plots on the right show chromosome 15 with the variant origin shown in red or blue dots. (E) VAF of the EFL1 variant in multiple tissue samples from I-1. (F) Genetic process underlying selection of EFL1p.Thr1069Ala cells in the patients. BMT, bone marrow transplantation; gDNA, genomic DNA; SNP, single-nucleotide polymorphism.
EFL1 deficiency impairs 80S ribosome assembly
Our interpretation of the genetic analysis assumes that the EFL1 variants are pathogenic and harbor a gradient of severity (Figure 2F). More specifically, p.His30Arg may possess a comparable severity with the frameshift variant (p.Gly827TrpfsTer13), and these 2 are more severe than p.Thr1069Ala. To test this, we measured ribosome assembly function in the presence of the EFL1 variants, because EFL1 is known to mediate GTP hydrolysis-coupled release of eIF6 together with SBDS during the maturation of the 60S ribosome subunit.7,15,16 Therefore, we monitored the ribosomal assembly status of the wild-type, small interfering RNA–mediated, or CRISPR/Cas9-mediated ablation of EFL1 (EFL1KD or EFL1−/−) in HeLa and K562 cell lines to further elucidate the molecular function of the mutant protein (Figure 3A; supplemental Figure 13). Polysome profiling of the EFL1KD and EFL1−/− cells showed a significantly reduced 80S peak (Figure 3A-C; supplemental Figure 13). This abnormal polysome profile was completely rescued after the introduction of FLAG-tagged wild-type EFL1 but not by the clones that harbored the mutations (Figure 3A-C; supplemental Figure 14). Interestingly, EFL1p.His30Arg failed to rescue the mutant phenotype, whereas EFL1p.Thr1069Ala displayed a moderate effect on ribosome assembly. These results indicate that EFL1 plays a crucial role in ribosome assembly, and EFL1p.His30Arg possesses a null function, whereas EFL1p.Thr1069Ala is hypomorphic.
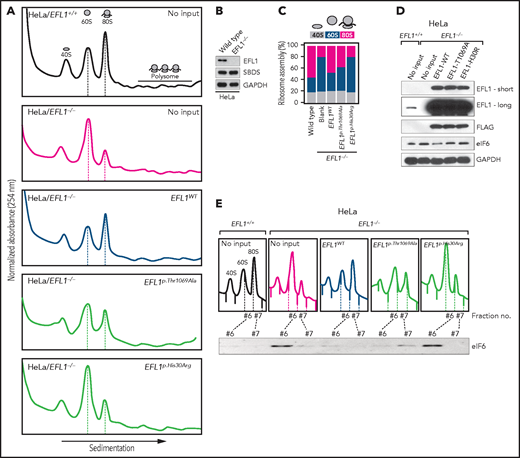
Nonsynonymous variants in EFL1 disrupt 80S assembly with varying severity. (A) Polysome profiling results of EFL1+/+ and EFL1−/− cells followed by transfection of vectors containing EFL1WT, EFL1p.Thr1069Ala, or EFL1p.His30Arg (as indicated in the top right corner of each plot). Note the different heights of 60S and 80S peaks in each experiment (dotted lines). (B) Immunoblot showing the CRISPR knockout of EFL1 in HeLa cells. (C) Quantification of the polysome profiling results displaying the relative occupancy of different ribosome statuses. Each bar corresponds to each experiment in panel B. (D) Immunoblots of EFL1 and eIF6 in wild-type or EFL1−/− HeLa cells with or without transfection of vectors containing EFL1WT, EFL1p.Thr1069Ala, or EFL1p.His30Arg. (E) Immunoblots of eIF6 from ribosomal fractions that best represent 60S and 80S of HeLa cells; each corresponds to the polysome profiles shown above. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Nonsynonymous variants in EFL1 disrupt 80S assembly with varying severity. (A) Polysome profiling results of EFL1+/+ and EFL1−/− cells followed by transfection of vectors containing EFL1WT, EFL1p.Thr1069Ala, or EFL1p.His30Arg (as indicated in the top right corner of each plot). Note the different heights of 60S and 80S peaks in each experiment (dotted lines). (B) Immunoblot showing the CRISPR knockout of EFL1 in HeLa cells. (C) Quantification of the polysome profiling results displaying the relative occupancy of different ribosome statuses. Each bar corresponds to each experiment in panel B. (D) Immunoblots of EFL1 and eIF6 in wild-type or EFL1−/− HeLa cells with or without transfection of vectors containing EFL1WT, EFL1p.Thr1069Ala, or EFL1p.His30Arg. (E) Immunoblots of eIF6 from ribosomal fractions that best represent 60S and 80S of HeLa cells; each corresponds to the polysome profiles shown above. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Molecular mechanism of EFL1-mediated SDS pathogenesis
Next, we explored the molecular function of the variants in ribosome assembly. Variant function was not mediated via phosphorylation of Thr1069, aberrant subcellular localization of EFL1, or changes in binding affinity to SBDS (supplemental Figure 15). Next, because the release of eIF6 from 60S is a crucial step for 80S assembly and is mediated by SBDS-EFL1, we investigated eIF6 level changes by altering EFL1. The assessment of eIF6 in wild-type and EFL1−/− cells revealed that the absence of EFL1 induced eIF6 levels, which was partially rescued by the introduction of EFL1p.Thr1069Ala or EFL1p.His30Arg (Figure 3D; supplemental Figure 16). Immunoblot analysis of each ribosomal subunit–bound fraction revealed that eIF6 was more highly enriched in the 60S ribosome fraction of the EFL1−/− cells as compared with the wild-type or EFL1-overexpressed cells (Figure 3E). Remarkably, introduction of EFL1p.Thr1069Ala rescued the increased eIF6 in the 60S fraction, whereas EFL1p.His30Arg failed to do so (Figure 3E). Also, absence of EFL1 caused an impaired shuttling of cytoplasmic eIF6 back to the nucleus, consistent with the previous observation (supplemental Figure 17).15 This result, along with the observation that changes in eIF6 and SBDS were not due to transcription levels (supplemental Figure 18), implies that the blocked exclusion of eIF6 and SBDS from the 60S ribosomal subunit is a mechanism by which the mutant protein functions, which results in impaired 80S ribosome assembly. This result also supports our hypothesis that the severity of p.His30Arg is greater than that of p.Thr1069Ala.
Deficiency of EFL1 orthologs reproduces SDS phenotypes in zebrafish and mouse models
To determine whether the milder allele (EFL1p.Thr1069Ala) was still pathogenic enough to cause SDS, a morpholino-targeting zebrafish model of efl1 was subjected to rescue experiments (supplemental Figure 19). The efl1 morphants had smaller heads and eyes as well as slightly bent tails and displayed an increased number of apoptotic cells during development (Figure 4A,C). Also, primitive erythrocytes and granulocytes were significantly reduced in the efl1 morphants, indicating impaired primitive hematopoiesis in these embryos (Figure 4B,D-E). All phenotypes were rescued by the introduction of wild-type human EFL1 mRNA, but less so by EFL1p.Thr1069Ala mRNA (Figure 4C-E), confirming that the milder efl1 allele caused SDS-like features in zebrafish.
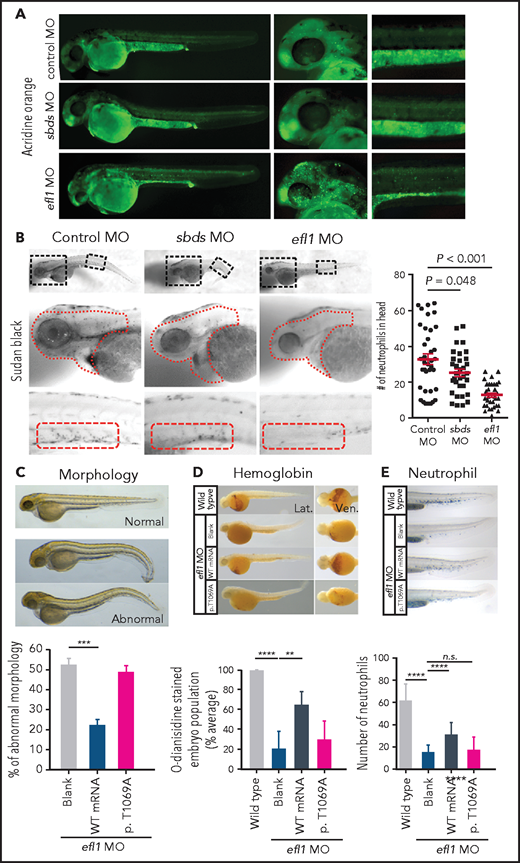
Zebrafish efl1 morphants model human SDS features. (A-B) Cell death (A) and neutrophil production (B) of zebrafish embryos 48 hours post fertilization by the knockdown of efl1 and sbds. (C-E) Rescue of efl1 MO animals using human EFL1WT or EFL1p.Thr1069Ala RNA, documenting morphology (C), hemoglobin production (D), and neutrophil production (E). **P < .01, ***P < .0005, ****P < .0001. MO, morpholino; n.s., not significant; WT, wild type.
Zebrafish efl1 morphants model human SDS features. (A-B) Cell death (A) and neutrophil production (B) of zebrafish embryos 48 hours post fertilization by the knockdown of efl1 and sbds. (C-E) Rescue of efl1 MO animals using human EFL1WT or EFL1p.Thr1069Ala RNA, documenting morphology (C), hemoglobin production (D), and neutrophil production (E). **P < .01, ***P < .0005, ****P < .0001. MO, morpholino; n.s., not significant; WT, wild type.
In addition, mouse models were created to further investigate the impact of Efl1 dysfunction. Efl1 knockout and p.Thr1076Ala knock-in mice (mouse Thr1076 is orthologous to human Thr1069; herein designated as Efl1m) were generated and subjected to phenotypic analyses (supplemental Figure 20). Embryos that were homozygous for the null allele (Efl1−/−) were not retrieved on embryonic day 8.5, implying the essential requirement for the gene in early embryogenesis (supplemental Figure 20). In contrast, mice homozygous for the knock-in allele (Efl1m/m) were viable and healthy (supplemental Figure 20), indicating a differential phenotypic tolerance between mice and humans. To model an accurate Efl1 dose that may induce an SDS-like phenotypic expression, we intercrossed the 2 strains and compared phenotypes of the CH animals (Efl1m/−) with the littermates (Efl1+/+, Efl1+/−, and Efl1+/m), with an emphasis on major SDS symptoms. The CH mice were smaller (Figure 5A-B) and died earlier (Figure 5C). The blood counts revealed reduced hemoglobin, white blood cells, and platelets (Figure 5D), and the bone marrow images displayed a consistent deficiency (Figure 5E). These results validated our observation that the reduced function of the EFL1p.Thr1069Ala variant caused SDS in human patients. To determine whether EFL1 expression was affected by the Efl1m variant, we measured the protein expression from embryonic day–17.5 livers (Figure 5F). The liver heterozygous for Efl1m (Efl1+/m) showed reduced EFL1 expression, suggesting that the variant not only reduced EFL1 activity but also destabilized the protein. This observation was consistent with the result from patient tissue (supplemental Figure 21). The expression of eIF6 was also detected in the CH livers and was increased compared with the wild-type livers, which was consistent with our previous observation in the HeLa cells (Figure 5G). Ribosome profiling of the wild-type and CH livers revealed that the CH livers showed a lower 80S peak compared with the wild-type livers (Figure 5H-I).
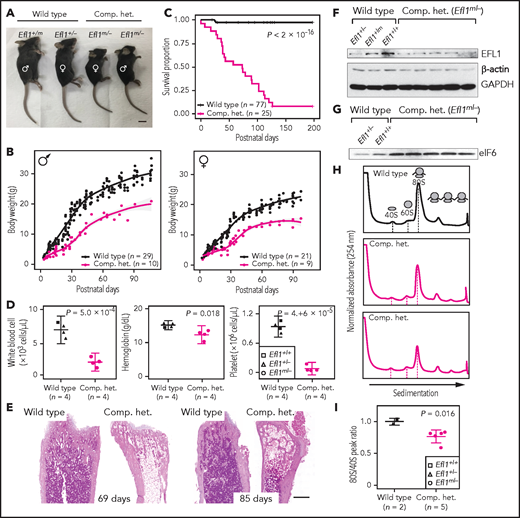
Mouse Efl1 mutants model human SDS features. (A) Body sizes of the CH mice (Efl1m/−) at 3 weeks compared with the wild-type littermates. (B) Body weight curves of the CH animals compared with the wild-type animals. Males (left) and females (right) are plotted separately. (C) Survival plot. (D) Hematopoietic features of the CH animals, measured at postnatal days 35-42. (E) Hematoxylin and eosin staining of the bone marrow from 69- and 85-day-old femurs. (F) Level of EFL1 protein in wild-type or CH livers. (G) Immunoblots of eIF6 from wild-type or CH livers. (H) Polysome profiling results of wild-type or CH mouse livers. (I) Quantification of the polysome profiling results. Scale bars, 1 cm (A); 0.5 mm (E).
Mouse Efl1 mutants model human SDS features. (A) Body sizes of the CH mice (Efl1m/−) at 3 weeks compared with the wild-type littermates. (B) Body weight curves of the CH animals compared with the wild-type animals. Males (left) and females (right) are plotted separately. (C) Survival plot. (D) Hematopoietic features of the CH animals, measured at postnatal days 35-42. (E) Hematoxylin and eosin staining of the bone marrow from 69- and 85-day-old femurs. (F) Level of EFL1 protein in wild-type or CH livers. (G) Immunoblots of eIF6 from wild-type or CH livers. (H) Polysome profiling results of wild-type or CH mouse livers. (I) Quantification of the polysome profiling results. Scale bars, 1 cm (A); 0.5 mm (E).
Screening downstream factors of EFL1 dysfunction
To further investigate the features of downstream genes that are strongly affected by reduced EFL1 function, we performed RNA sequencing on the total RNA and 40S-, 80S-, and polysome-bound RNAs from wild-type and EFL1−/− K562 cells (Figure 6A; supplemental Figure 22). The 60S-bound RNAs were not analyzed, because it is unlikely that the large subunit would bind with RNA by itself. The 248 genes that were decreased in the 80S-bound fraction compared with the 40S-bound fraction in the mutant cells were extracted. A gene ontology analysis suggested that RP genes (GO:0003735) were the major constituents of the genes that were reduced by the absence of EFL1 (adjusted P = 5.2 × 10−77; adjusted by the Benjamini-Hochberg method; Figure 6B-C). There was no significantly enriched gene group in the increased gene set. The fractions of the transcripts bound by 40S, 80S, or polysome differed substantially for the RP genes in the absence of EFL1 (P < 1.0 × 10−13 for differences in both 40S- and 80S-bound transcripts; Wilcoxon signed rank test), whereas neither the TP53 target genes nor other genes showed such a change (Figure 6D; P > .05; Wilcoxon signed rank test). The enrichment of RP genes in 40S was rather unexpected; we infer that it may reflect molecular congestion resulting from the lack of EFL1, causing a delay of the RP transcripts in progression to the 80S ribosome–bound form. Next, we compared whether transcript sizes (supplemental Figure 23), expression levels, or consensus sequence elements in the 5' untranslated regions may serve as factors that enabled RP-specific regulation. Notably, highly expressed RP transcripts with a TOP element (5'-CUUYCUUUUNS-3') were specifically altered (Figure 6E; supplemental Figure 24; P = 6.9 × 10−7). This result revealed that, among all the genes in the genome, 80S ribosome assembly of RP transcripts containing a TOP element was heavily dependent on normal EFL1 function.
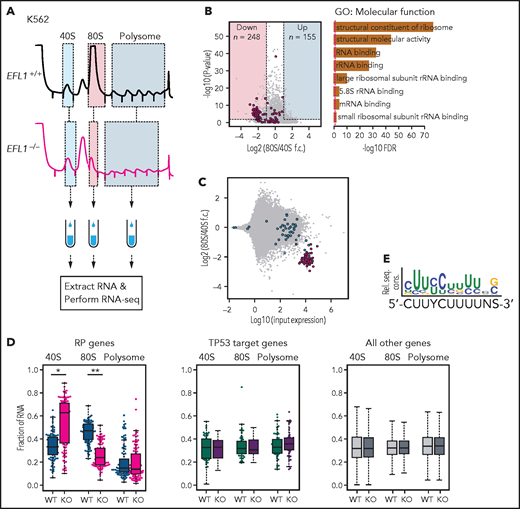
EFL1 is required for ribosomal protein (RP) synthesis. (A) Schematic diagram of the RNA expression analysis from K562 ribosomal fractions. (B) Volcano plot of the genes plotting differential enrichments of wild-type expression vs mutant expression in the 80S fraction compared with the 40S fraction (left). GO analysis of the 248 downregulated genes (right). Red dots depict RP genes (GO:0022625 and GO:0022627; n = 118). (C) Fold changes of 80S-enriched genes normalized by 40S, depicted against transcriptome. Red indicates significantly downregulated RP genes in panel B; blue indicates other RP genes. (D) Fractions of 40S-, 80S-, or polysome-bound RNAs for RP genes (n = 108; left), TP53 target genes (n = 51; middle), and all other genes (n = 11 812; right) are displayed. (E) Consensus sequence profile of downregulated RP gene 5' untranslated regions, deducing a terminal oligopyrimidine (TOP) sequence. *P = 4.7 × 10−14, **P = 1.5 × 10−26 by Wilcoxon signed rank test; all other test P values >0.05. KO, knockout; rel. seq. cons., relative sequence conservation from the MEME run; RNA-seq, RNA sequencing; rRNA, ribosomal RNA; WT, wild type.
EFL1 is required for ribosomal protein (RP) synthesis. (A) Schematic diagram of the RNA expression analysis from K562 ribosomal fractions. (B) Volcano plot of the genes plotting differential enrichments of wild-type expression vs mutant expression in the 80S fraction compared with the 40S fraction (left). GO analysis of the 248 downregulated genes (right). Red dots depict RP genes (GO:0022625 and GO:0022627; n = 118). (C) Fold changes of 80S-enriched genes normalized by 40S, depicted against transcriptome. Red indicates significantly downregulated RP genes in panel B; blue indicates other RP genes. (D) Fractions of 40S-, 80S-, or polysome-bound RNAs for RP genes (n = 108; left), TP53 target genes (n = 51; middle), and all other genes (n = 11 812; right) are displayed. (E) Consensus sequence profile of downregulated RP gene 5' untranslated regions, deducing a terminal oligopyrimidine (TOP) sequence. *P = 4.7 × 10−14, **P = 1.5 × 10−26 by Wilcoxon signed rank test; all other test P values >0.05. KO, knockout; rel. seq. cons., relative sequence conservation from the MEME run; RNA-seq, RNA sequencing; rRNA, ribosomal RNA; WT, wild type.
Discussion
We identify and describe a unique bone marrow–specific somatic UPD event that preferentially selects cells with EFL1 alleles of a weaker severity. Although biallelic variants in EFL1 may occur in other parts of the body, the hematologic system was partially rescued by a somatic UPD resulting in homozygous EFL1p.Thr1069Ala with a weaker severity. We provide evidence suggesting that a majority of the cells carrying homozygous EFL1p.Thr1069Ala are pathogenic. We hypothesize that a somatic UPD in the patients could occur and be detected because of the dynamic nature of bone marrow, where the whole stem cell population can be potentially replaced by a few clones. Indeed, the somatic UPD was not detected in other solid organs or tissues that were available for investigation. Two lines of evidence suggest infrequent LOH events in the EFL1 locus in normal populations (2.7 × 10−4 to 7.7 × 10−4; see “Results”). The odds of 1 person having all the p.Thr1069Ala and p.His30Arg variants and the LOH by UPD in the EFL1 locus by chance is roughly estimated as 1 in 3 trillion, indicating that detectable UPD is not an independent event but is associated with the biallelic EFL1 variants. It is still not clear if the EFL1 dysfunction somehow contributed to the occurrence of UPD, causing the resulting clone to be expanded by positive selection. Nevertheless, the degree of mosaicism did not seem to directly determine clinical severity (supplemental Figure 12). More accurate temporal and spatial occurrence of the event could be delineated if additional clinical samples were to become available.
Several lines of evidence from our experiments suggest that although p.Thr1069Ala is the mildest among the variants that we found, it is still functional and hypomorphic: (1) the parents of patients who were heterozygous for EFL1p.Thr1069Ala were asymptomatic, whereas the patients carried the variant in a homozygous status in their bone marrow and showed the pathology; (2) a ribosome profiling assay using a variant allele partially rescued the 80S assembly problem, whereas the wild-type allele completely rescued it (Figure 3); (3) efl1 morpholino-treated zebrafish were partially rescued by the variant-containing RNA (Figure 4); and (4) Efl1m/− mice displayed an SDS-like phenotype, which was of intermediate severity relative to the Efl1−/− and Efl1+/− mice (Figure 5). It is notable that although our mouse model successfully phenocopied most of the SDS features, the genotype was not in complete concordance with that of human patients. Our mouse model with Efl1m/m did not show significant phenotypes, whereas human patients with bone marrow–specific homozygous EFL1p.Thr1069Ala by somatic UPD still had hematologic abnormalities, such as anemia and neutropenia. This discrepancy between the 2 species is not rare,26,27 and it again underscores differences in tolerance of a given variant, perhaps resulting from different physiologic and genetic systems, which is critical in modeling human clinical features in mice.
Nonetheless, we demonstrated that the altered function of EFL1 specifically influenced the translation of RP genes containing a TOP element in the 5' untranslated region, which led to a mechanistic mimicry of Diamond-Blackfan anemia (DBA), which is another ribosomopathy caused by insufficient RP doses.28,29 The activation of TP53 is considered a targetable downstream pathway that leads to SDS or a DBA phenotype.30-32 However, our data suggest that the loss of EFL1 does not induce TP53 activation, which is consistent with previous studies of the zebrafish sbds model and DBA (Figure 6D; supplemental Figure 18 [RNA sequencing of EFL1−/− cells]; supplemental Figure 21 [western blots of patient tissue and EFL1−/− cells]).33,34 In addition to the TP53 pathway, an extended search for expression changes in other potentially SDS causal genes did not reveal significant differences (supplemental Figures 18 and 19).
It is known that LARP1 directly binds to the TOP element of RP genes to repress translation in a phosphorylation-dependent manner and that mTOR partially regulates LARP1 phosphorylation.35,36 To determine if we could use this pathway to derepress RP translation and rescue the SDS phenotype in the animal models, we considered a molecular signaling pathway that may regulate RP gene translation through the TOP element. However, both LARP1 binding to RP TOP elements and mTOR signaling were unchanged in the mutant cells, suggesting an alternative mechanism that may regulate RP translation (data not shown).
Here, we demonstrate a mechanism by which biallelic variants of EFL1 phenocopied classical SDS in 3 unrelated patients. The bone marrow–specific somatic UPD in these patients mitigated the potentially catastrophic hematologic phenotype by homozygosing the less damaging variant (EFL1p.Thr1069Ala). We demonstrate that defective EFL1 caused impaired 80S ribosomal assembly and that the zebrafish and mouse models displayed similar features to humans through the alteration of 80S ribosome assembly of RP transcripts. An extensive search of such SDS patients may provide more insight into the development of somatic mosaicism and subsequent molecular cascades that may lead to new avenues of treatment of ribosomopathy.
Acknowledgments
The authors thank the patients and families who participated in this study; Jung-Ah Kim and Hyoung-Jin Kim at Seoul National University Hospital and Cho-Rong Lee and Eunha Kim at Seoul National University for collecting and interpreting hematologic data and interpreting images; and the University of Utah Mutation Generation and Detection Core for development of the Efl1 knock-in mice. O.K. acknowledges the computing resources provided by Bebop, a high-performance computing cluster operated by the Laboratory Computing Resource Center at Argonne National Laboratory.
This study was partly supported by grants 2014M3C9A2064686 and 2019R1A2C2010789 (M.C.) and 2017R1A2A2A05069691 (H.H.K.) from the National Research Foundation of Korea, and awards REU CHE-1062629 and RUI MCB-1607024 (N.T.W.) from the National Science Foundation. This research was partly supported by the Korea Mouse Phenotyping Project (2013M3A9D5072550) of the Ministry of Science, ICT and Future Planning through the National Research Foundation.
Authorship
Contribution: M.C., H.J.K., and H.H.K. conceived the study. S.L. and M.C. performed genetic analysis and statistical evaluations; C.H.S., S.L., S.D.J., H.H.K., J.L., and M.C. performed molecular biology and biochemistry experiments and assessed the results; J.L., S.L., S.J.S., M.C., and J.K.S. performed mouse experiments; J.-D.K., A.-R.K., B.P., and S.-W.J. performed zebrafish experiments; C.R.H., J.S.K., Y.B.S., O.-H.K., J.M.K., T.-J.C., and H.J.K. provided patient care and generated clinical data; N.T.W., O.K., and T.Y. analyzed protein structure. M.C., S.L., C.H.S., C.R.H., J.L., H.J.K., and H.H.K. wrote the manuscript; and all authors approved the final version of the manuscript.
The current affiliation for S.L. is Department of Neurosciences, University of California San Diego, La Jolla, CA.
The current affiliation for C.H.S. is Laboratory of Genetics and Genomics, National Institute on Aging Intramural Research Program, National Institutes of Health, Baltimore, MD.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Murim Choi, Department of Biomedical Sciences, Seoul National University College of Medicine, Seoul, South Korea; e-mail: murimchoi@snu.ac.kr; Hyeon Ho Kim, Department of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology, Sungkyunkwan University, Seoul, South Korea; e-mail: hyeonhkim@skku.edu; and Hyoung Jin Kang, Department of Pediatrics, Seoul National University College of Medicine, Seoul, South Korea; e-mail: kanghj@snu.ac.kr.
The raw sequencing data discussed in this publication have been deposited in the Korean Nucleotide Archive (accession #PRJKA210036).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal