

Key Points
Genetic knockout of PRDM1 in antitumor T cells enhances persistence and therapeutic response in multiple adoptive immunotherapy models.
PRDM1-knockout T cells acquire gene expression profiles of early memory T cells through genome-wide epigenetic changes.

Abstract
Adoptive cancer immunotherapy can induce objective clinical efficacy in patients with advanced cancer; however, a sustained response is achieved in a minority of cases. The persistence of infused T cells is an essential determinant of a durable therapeutic response. Antitumor T cells undergo a genome-wide remodeling of the epigenetic architecture upon repeated antigen encounters, which inevitably induces progressive T-cell differentiation and the loss of longevity. In this study, we identified PR domain zinc finger protein 1 (PRDM1) ie, Blimp-1, as a key epigenetic gene associated with terminal T-cell differentiation. The genetic knockout of PRDM1 by clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) supported the maintenance of an early memory phenotype and polyfunctional cytokine secretion in repeatedly stimulated chimeric antigen receptor (CAR)-engineered T cells. PRDM1 disruption promoted the expansion of less differentiated memory CAR-T cells in vivo, which enhanced T-cell persistence and improved therapeutic efficacy in multiple tumor models. Mechanistically, PRDM1-ablated T cells displayed enhanced chromatin accessibility of the genes that regulate memory formation, thereby leading to the acquisition of gene expression profiles representative of early memory T cells. PRDM1 knockout also facilitated maintaining an early memory phenotype and cytokine polyfunctionality in T-cell receptor-engineered T cells as well as tumor-infiltrating lymphocytes. In other words, targeting PRDM1 enabled the generation of superior antitumor T cells, which is potentially applicable to a wide range of adoptive cancer immunotherapies.
Introduction
The adoptive transfer of T cells that recognize tumor antigens is a potentially curative therapeutic approach for advanced cancer, as exemplified by the marked clinical efficacy of CD19-targeting chimeric antigen receptor (CAR)-T cell therapy against B-cell malignancies. However, a substantial number of patients treated with the CD19 CAR-T cells experience relapse even after complete remission.1-5 Moreover, durable therapeutic response has rarely been achieved in CAR-T cell therapies targeting solid tumors.6-8 The loss of tumor antigen expression,1,8-10 inefficient trafficking of infused T cells into the tumor site,11,12 and immunosuppressive tumor microenvironments13,14 are the major obstacles that result in treatment failure. Furthermore, the long-term persistence of infused tumor-specific T cells, including CAR-T cells, T-cell receptor (TCR)-T cells, and tumor-infiltrating lymphocytes (TIL), is essential for sustained clinical response.4,15,16 Antitumor T cells prepared in vitro acquire a phenotype of memory T cells with long-term survival potential.17 Accumulating evidence suggests that the transient modulation of specific signaling pathways enables the suppression of excessive T-cell differentiation and the loss of a memory phenotype during in vitro expansion.18-23 However, T cells rapidly undergo differentiation and exhaustion upon repeated antigen exposure in vivo, which hinders durable antitumor response.24,25
Terminally differentiated T cells possess distinct epigenetic and transcriptional landscapes compared with naïve and memory T cells, which likely underlie dysfunctional T cell properties such as poor survival and impaired cytokine secretion.26-30 Immune checkpoint blockade transiently reactivates the effector functions of terminally differentiated and exhausted T cells; however, it does not fundamentally affect their epigenetic architecture. In this study, we hypothesized that the genetic modulation of key regulators orchestrating the epigenetic profiles in terminally differentiated T cells may halt or suppress progressive T-cell differentiation, thereby enhancing the survival of adoptively transferred T cells. We demonstrated that PRDM1 (Blimp-1) is an essential epigenetic factor related to terminal T-cell differentiation, and its genetic ablation in antitumor T cells augments durable antitumor responses in multiple adoptive immunotherapy models.
Methods
In vitro culture of human T cells
Healthy donor-derived peripheral blood mononuclear cells (PBMCs) were stimulated with mitomycin C-treated K562 cells expressing anti-CD3 mAb (clone OKT3)-derived single-chain variable fragment (scFV) on the cell surface as well as CD80 (K562-OKT3/CD80) at an effector to target (E:T) ratio of 7:1. We cultured the stimulated T cells in the presence of recombinant IL-2 (100 IU/mL; PeproTech). Retroviral transduction of the T cells was performed 2 days following their stimulation using RetroNectin (Takara Bio). We restimulated the CD19-targeting CAR-T cells by K562 cells transduced with CD19 or NALM6 at an E:T ratio of 5:1 or 1:1, respectively. Mesothelin-targeting CAR-T cells were restimulated by K562 cells expressing mesothelin at an E:T ratio of 5:1. The DMF5 TCR-transduced T cells were stimulated by coculture with irradiated T2 cells, loaded with 1 μg/mL of MART127-35 peptide at an E:T ratio of 5:1.
CRISPR/Cas9-mediated knockout of epigenetic genes
Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-mediated gene knockout was performed by the electroporation of a ribonucleoprotein (RNP) complex composed of Cas9 protein and guide RNA using the NEPA 21 electroporator (Nepa Gene). Detailed protocols are described in the supplemental Methods, available on the Blood Web site. Genomic DNA was extracted 48 hours after electroporation by using NucleoSpin DNA Rapid Lyse (Macherey Nagel), and the regions surrounding the target sequences were amplified by PCR and submitted to Sanger Sequencing. The knockout efficiency was estimated based on the sequencing data using the Inference of CRISPR Edits (ICE) analysis.31
Flow cytometry analysis
Flow cytometry analysis was performed with a BD LSRFortessa (BD Biosciences) using the antibodies listed in supplemental Table 1. For cytokine production analysis, we cocultured the T cells with target cells for 6 hours. Brefeldin A (BioLegend) was added to the cultures 2 hours after stimulation. Subsequently, the cells were fixed and permeabilized using a Cyto-Fast Fix/Perm kit (BioLegend), followed by intracellular staining. For a flow cytometry analysis of TCF7 expression, we fixed the T cells with 4% paraformaldehyde and permeabilized them with ice-cold methanol. Data analysis was performed using FlowJo software (version 10.7.1; BD Biosciences).
Mice
We used 4- to 10-week-old male NSG mice (Charles River Laboratories) to analyze human T-cell functions in vivo. In each experiment, mice were monitored for their overall health status and body weight at least thrice a week and sacrificed upon exhibiting 1 of the following symptoms: >20% loss of initial body weight, pronounced lethargy, severe diarrhea, hunched posture, and severe dermatitis. In the NALM6 leukemia model, we analyzed the tumor burden using IVIS Lumina II (Perkin Elmer). For analyzing leukemia-related survival, mice were censored upon meeting the above-mentioned criteria owing to an expansion of the infused T cells. The cause of death was determined based on spleen analysis by flow cytometry, IVIS, and PB analysis data. In the A375 model, progression-free survival was defined as the time to tumor regrowth exceeding 20 mm3 at 2 consecutive time points after tumor rechallenge. Detailed experimental protocols are described in the supplemental Methods.
In vitro cytotoxicity assay
To evaluate the cytotoxic activity of CAR-T cells, we cocultured 1 × 105 CAR-T cells with the indicated ratio of EGFP+ target cells. The absolute counts of viable target cells were determined by flow cytometry. The frequency of surviving tumor cells was calculated as the ratio of the cell counts incubated without CAR-T cells. Moreover, dead cells were discriminated using the LIVE/DEAD Fixable Near-IR Dead Cell Stain Kit (Thermo Fisher Scientific).
RNA-sequencing and ATAC-sequencing analysis
The CD19 CAR-T cells derived from 3 different donors were electroporated with Cas9/sgRNAs targeting PRDM1 and stimulated thrice with NALM-6 every 3 to 4 days. The CD8+ CAR-T cells were isolated by flow cytometry and subjected to RNA sequencing (Takara Bio) and assay for transposase-accessible chromatin with high-throughput (ATAC)-sequencing analysis (GENEWIZ) (please refer to the supplemental Methods for the detailed analysis pipeline).
Immunoblotting
The following antibodies were used for the immunoblot analysis: anti-Blimp1 (#9115; Cell Signaling Technology), anti-TOX/TOX2 (#36778; Cell Signaling Technology), anti-phospho-STAT1 (Tyr701) (#9167; Cell Signaling Technology), anti-STAT1 (#9172; Cell Signaling Technology), anti-β-actin (sc-47778; Santa Cruz Biotechnology), HRP-conjugated goat anti-mouse IgG (H + L) (#4021; Promega), and anti-rabbit IgG (#4011; Promega).
TIL culture
Tumor specimens derived from patients with lung, ovarian, endometrial, and cervical cancer were minced and cultured with RPMI GlutaMAX (Gibco) containing 10% fetal bovine serum (FBS), 3000 IU/mL recombinant human IL-2 (Proleukin; Novartis), and sodium pyruvate (Gibco) for 14 days. We stimulated the expanded TILs with K562-OKT3/41BBL (E:T ratio of 10) and ablated them with PRDM1 4 days later. For the direct ex vivo analysis of the TIL, tumor specimens were dissociated into single-cell suspensions using gentleMACS Dissociators (Miltenyi Biotech).
Statistical analysis
Statistically significant differences between the 2 groups were assessed using a 2-tailed paired or unpaired Student t test for parametric data. The log-transformed values were compared depending on the nature of the data. We performed the Mann-Whitney U test for nonparametric data that were not corrected by log transformation. More than 2 groups were compared using ordinary or repeated measures one-way analysis of variance (ANOVA) with multiple comparisons test. Differences were considered statistically significant for P values <.05. In the mice experiments, the Kaplan-Meier curve was analyzed using the log-rank test. All statistical analyses were performed using GraphPad Prism software (version 9.0.0).
Study approval
This study was conducted in accordance with the tenets of the Declaration of Helsinki, and was approved by the Research Ethics Board of the Aichi Cancer Center, Nagoya, Japan. Written informed consent was obtained from all patients who provided the TIL samples. All animal experiments were approved by the Animal Care and Use Committee of the Aichi Cancer Center Research Institute (Nagoya, Japan).
Results
PRDM1 knockout suppresses terminal differentiation in repeatedly stimulated T cells
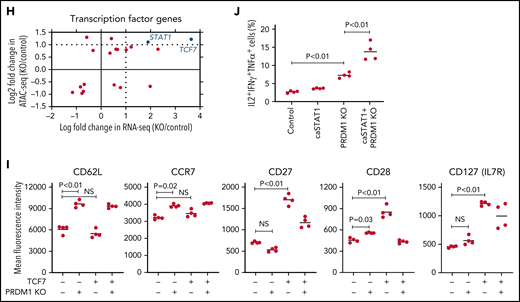
To recapitulate the tumor microenvironment that continuously exposes antitumor T cells to the tumor antigen, we repeatedly stimulated T cells engineered with a CD19-targeting CAR gene encoding the clone FMC63-derived scFV linked with CD28 and CD3ζ signaling domains by CD19+ target cells (K562-CD19 or NALM6) at different intervals (supplemental Figure 1A,B). The expanded CAR-T cells nearly lost a CD45RA+CD62L+CCR7+CD27+CD28+ T-cell population (stem cell-like memory T cells: TSCM) and included a small population of CD45RA-CD62L+CCR7+CD27+CD28+ cells, representing a central memory phenotype (TCM) (supplemental Figure 1C-F). The CAR-T cells stimulated by K562-CD19 expressed higher levels of PD1, LAG3, and TIM3 than those stimulated by NALM6, likely because of the high antigen load (supplemental Figure 1G-H). Accompanied by terminal differentiation, repeatedly stimulated CAR-T cells lost their polyfunctional cytokine production capacity (supplemental Figure 1I-K). Using this experimental setting, we investigated if modulation of the specific epigenetic factors can halt terminal T-cell differentiation and impaired cytokine production. PRDM1, which encodes Blimp1, is one of the epigenetic factors associated with the development of short-lived effector T cells.32,33Prdm1 knockout in T cells promoted memory formation in virus infection mouse models.34,35 To investigate if the knockout of PRDM1 modifies CAR-T cell properties, the CD19 CAR-T cells were genetically ablated with PRDM1 using CRISPR/Cas9 and subsequently underwent repeated stimulation (Figure 1A). The efficient loss of Blimp1 expression was confirmed at protein levels in PRDM1-knockout T cells (Figure 1B). We found that the knockout of PRDM1 improved the maintenance of TSCM and TCM phenotypes in repeatedly stimulated CAR-T cells without affecting T-cell proliferation (Figure 1C-E). Furthermore, we analyzed the expression of another canonical memory marker, IL7R. PRDM1-ablated T cells displayed a higher frequency of CD45RA+/−CD62L+CCR7+CD27+CD28+IL7R+ cells compared with the control (Figure 1F). There was no significant change in the PRDM1 knockout frequency after repeated stimulation, thereby suggesting PRDM1 deletion did not impair T-cell proliferation (Figure 1G).

PRDM1 knockout maintains an early memory phenotype in CD19-directed CAR-T cells. (A) CD19-targeting CAR-T cells were electroporated with a Cas9/sgRNA ribonucleoprotein (RNP) complex against PRDM1 or electroporated without RNP (no target). The CAR-T cells were then stimulated thrice by K562-CD19 or NALM6. (B) Immunoblotting of Blimp-1 in control or PRDM1-knockout CAR-T cells generated from 3 different donors. (C) Fold expansion of T cells during the 3 stimulations (n = 11, paired 2-tailed Student t test). (D-F) Memory markers were analyzed in the CD8+ CAR-T cell population after the third stimulation. The data shown are representative flow cytometry plots of primary CD8+ T cells or CAR-T cells stimulated by K562-CD19 (D) and the frequency of CD45RA+/−CD62L+CCR7+CD27+CD28+ cells (E) or CD45RA+/−CD62L+CCR7+CD27+CD28+IL7R+ cells (F) in the CD8+ CAR-T cell population (n = 12 or 11, paired 2-tailed Student t test for each). (G) Knockout efficiency was evaluated by Inference of CRISPR Edits (ICE) analysis using the genome extracted before and after repeated stimulation (n = 11, repeated measures one-way ANOVA with multiple comparisons test). The data were obtained from different donor samples. Horizontal lines denote the mean values. NS, not significant.
PRDM1 knockout maintains an early memory phenotype in CD19-directed CAR-T cells. (A) CD19-targeting CAR-T cells were electroporated with a Cas9/sgRNA ribonucleoprotein (RNP) complex against PRDM1 or electroporated without RNP (no target). The CAR-T cells were then stimulated thrice by K562-CD19 or NALM6. (B) Immunoblotting of Blimp-1 in control or PRDM1-knockout CAR-T cells generated from 3 different donors. (C) Fold expansion of T cells during the 3 stimulations (n = 11, paired 2-tailed Student t test). (D-F) Memory markers were analyzed in the CD8+ CAR-T cell population after the third stimulation. The data shown are representative flow cytometry plots of primary CD8+ T cells or CAR-T cells stimulated by K562-CD19 (D) and the frequency of CD45RA+/−CD62L+CCR7+CD27+CD28+ cells (E) or CD45RA+/−CD62L+CCR7+CD27+CD28+IL7R+ cells (F) in the CD8+ CAR-T cell population (n = 12 or 11, paired 2-tailed Student t test for each). (G) Knockout efficiency was evaluated by Inference of CRISPR Edits (ICE) analysis using the genome extracted before and after repeated stimulation (n = 11, repeated measures one-way ANOVA with multiple comparisons test). The data were obtained from different donor samples. Horizontal lines denote the mean values. NS, not significant.
We evaluated the exhaustion markers of restimulated CAR-T cells. Intriguingly, PRDM1-knockout CAR-T cells revealed slightly elevated expression of PD1 and decreased expression of TIM3 upon restimulation with K562-CD19 (Figure 2A-B). Consistent with PD1 upregulation, PRDM1-ablated CAR-T cells displayed elevated TOX expression, a transcriptional regulator of immunoinhibitory molecules, compared with the control CAR-T cells (Figure 2C).36-38 In other words, terminal T-cell differentiation and the induction of immunoinhibitory molecules were regulated by distinct transcriptional mechanisms. Despite PD1 upregulation, PRDM1-knockout T cells were superior in the production of multiple cytokines regardless of the stimulation protocol (Figure 2D-F). These results were reproduced upon individually electroporating CAR-T cells with 2 different guide RNAs targeting PRDM1 (supplemental Figure 2A-F). Moreover, CAR-T cells transduced with N-terminally truncated Blimp-1 (tBlimp1), which lacks a DNA-binding domain and functions in a dominant-negative manner in B cells,39 were superior in maintaining an early memory phenotype and polyfunctional cytokine production (supplemental Figure 3A-E). Our findings suggest that the observed effect of PRDM1 knockout indeed resulted from the loss of PRDM1 rather than from off-target effects. We also tested knockout of the DNA hydroxymethylase TET2 as a comparison, whose disruption enhanced CAR-T cell longevity in a clinical study.40 TET2 ablation also maintained T cells with an early memory phenotype significantly better than the control T cells (supplemental Figure 4A-E). PRDM1 knockout was more prominent in improving the maintenance of TSCM and TCM populations and cytokine polyfunctionality after repeated stimulation by K562-CD19 and NALM6. Moreover, PRDM1-knockout cells displayed improved expression of early memory T cell markers as well as cytokine production in the CD4+ CAR-T cell population (supplemental Figure 5A-C).
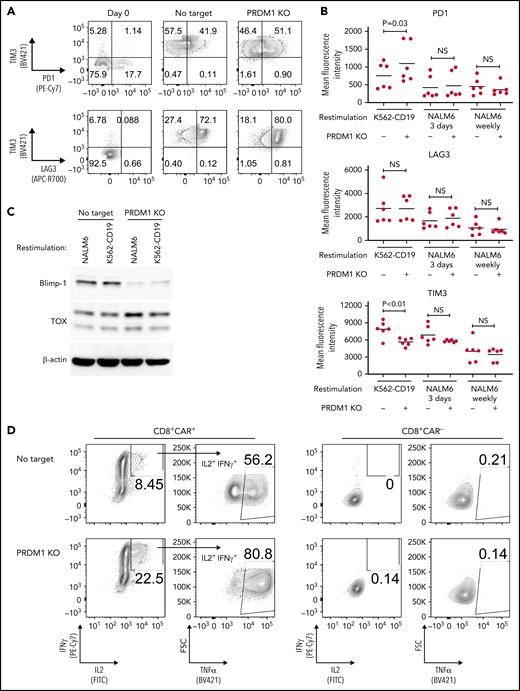
PRDM1-knockout CAR-T cells maintain polyfunctional cytokine production after repeated stimulation. (A-B) The expression of immunoinhibitory molecules was analyzed after the third restimulation. Representative flow cytometry plots of primary CD8+ T cells or CAR-T cells repeatedly stimulated by K562-CD19 (A) and mean fluorescence intensity of PD1, LAG3, and TIM3 in the CD8+ CAR-T cell population (B) (n = 6, paired 2-tailed Student t test). (C) Expression of TOX in repeatedly stimulated CAR-T cells with or without PRDM1 knockout was analyzed by immunoblotting. (D-F) Cytokine production after the third stimulation was analyzed by flow cytometry (IL2, IFNγ, and TNFα). Representative flow cytometry plots of CAR-T cells restimulated by K562-CD19 (D), pie charts of the percentages of CAR-T cells producing single, double, and triple cytokines (E), and the frequency of IL2+IFNγ+TNFα+ cells in the CD8+ CAR-T cell population are shown (F) (n = 11, paired 2-tailed Student t test). The data presented in B and F are derived from different donor samples. Horizontal lines indicate the mean values. NS, not significant.
PRDM1-knockout CAR-T cells maintain polyfunctional cytokine production after repeated stimulation. (A-B) The expression of immunoinhibitory molecules was analyzed after the third restimulation. Representative flow cytometry plots of primary CD8+ T cells or CAR-T cells repeatedly stimulated by K562-CD19 (A) and mean fluorescence intensity of PD1, LAG3, and TIM3 in the CD8+ CAR-T cell population (B) (n = 6, paired 2-tailed Student t test). (C) Expression of TOX in repeatedly stimulated CAR-T cells with or without PRDM1 knockout was analyzed by immunoblotting. (D-F) Cytokine production after the third stimulation was analyzed by flow cytometry (IL2, IFNγ, and TNFα). Representative flow cytometry plots of CAR-T cells restimulated by K562-CD19 (D), pie charts of the percentages of CAR-T cells producing single, double, and triple cytokines (E), and the frequency of IL2+IFNγ+TNFα+ cells in the CD8+ CAR-T cell population are shown (F) (n = 11, paired 2-tailed Student t test). The data presented in B and F are derived from different donor samples. Horizontal lines indicate the mean values. NS, not significant.
PRDM1-ablated CAR-T cells show superior persistence and antitumor effects in vivo
We investigated if PRDM1-disrupted CAR-T cells demonstrated functional superiority in vivo. On being adoptively transferred into irradiated tumor-free NSG mice, PRDM1-knockout CAR-T cells were detected at significantly higher frequencies in the PB (Figure 3A-B). Lethal xenogeneic graft-versus-host disease (GVHD) induced by the expanded T cells was more frequently developed in mice infused with the PRDM1-knockout CAR-T cells than that in controls (Figure 3C; supplemental Figure 6A-C). The PRDM1 knockout frequency in CAR-T cells that persisted in the spleen was significantly higher than that in infusion products, thus suggesting PRDM1-ablated CAR-T cell clones preferentially expanded in vivo (Figure 3D). Prolonged proliferative capacity and persistence in vivo are some of the hallmarks of immature memory T cells.41 Therefore, PRDM1-knockout CAR-T cells acquire functional and phenotypic features of early memory T cells.


PRDM1-ablated CD19-targeting CAR-T cells show superior persistence and antitumor activity in vivo. (A-C) CD19-targeting CAR-T cells electroporated with or without Cas9 and sgRNAs against PRDM1 were infused into tumor-free NSG mice. The frequency of human T cells in the PB was monitored at the indicated time points (B) (unpaired 2-tailed Student t test). Kaplan-Meier analysis for overall survival after CAR-T cell infusion (C) (n = 7 mice for each group, log-rank test). The data are a composite of 2 independent experiments. (D) The PRDM1 knockout efficiency in infusion products and the persisting CAR-T cells within the spleen was analyzed (n = 7, paired 2-tailed Student t-test). (E) Cytolytic activity of control or PRDM1-knockout CAR-T cells against the indicated target cells was analyzed with flow cytometry (n = 3 cultures, unpaired 2-tailed Student t test for each condition). Representative data of 2 experiments. (F-G) Cytolytic activity of control or PRDM1-knockout CAR-T cells against NALM6 (F) or K562-CD19 (G) at the indicated time points (n = 3-4 cultures, one-way ANOVA with multiple comparisons test). (H-L) NSG mice were intravenously infused with NALM6-GL, and then with CAR-T cells with or without PRDM1 knockout 10 days later (n = 10 mice for each CAR). (I) The frequency of human T cells in the PB was analyzed at the indicated time points (unpaired 2-tailed Student t test of the log-transformed values for each time point). (J) Total flux in the whole body was measured by in vivo bioluminescence imaging (unpaired 2-tailed Student t test of the log-transformed values). (K) Kaplan-Meier analysis for leukemia-related survival after NALM6-GL infusion (log-rank test). In (H-K), representative data of 2 independent experiments are shown. (L) The PRDM1-knockout frequency was compared between infusion products and T cells within the spleen of the mice that developed xenogeneic GVHD (n = 4, paired 2-tailed Student t test). (M-N) NALM6-GL-engrafted NSG mice were transplanted with control or PRDM1-knockout CAR-T cells generated in the same protocol as that shown in (H). The frequency of CD8+ or CD4+ CAR-T cells (M) (n = 6, unpaired 2-tailed Student t test of the log-transformed values) or those with TSCM and TCM phenotypes (N) (n = 6, unpaired 2-tailed Student t test for each time point) in the PB was determined by flow cytometry. (O-R) NSG mice subcutaneously inoculated with A375-CD19 (day 0) were treated with CAR-T cells with or without PRDM1 knockout (day 7). The mice were rechallenged with A375-CD19 (day 24) and monitored for tumor progression (n = 9 mice for each group). The data shown are serial monitoring of tumor volume (P), relapse-free survival (Q) (log-rank test), and the frequency of human T cells at the indicated time points (R) (unpaired 2-tailed Student t test of the log-transformed values). In B, E, F, G, I, J, M, N, and R, horizontal lines denote the mean values. NS, not significant.
PRDM1-ablated CD19-targeting CAR-T cells show superior persistence and antitumor activity in vivo. (A-C) CD19-targeting CAR-T cells electroporated with or without Cas9 and sgRNAs against PRDM1 were infused into tumor-free NSG mice. The frequency of human T cells in the PB was monitored at the indicated time points (B) (unpaired 2-tailed Student t test). Kaplan-Meier analysis for overall survival after CAR-T cell infusion (C) (n = 7 mice for each group, log-rank test). The data are a composite of 2 independent experiments. (D) The PRDM1 knockout efficiency in infusion products and the persisting CAR-T cells within the spleen was analyzed (n = 7, paired 2-tailed Student t-test). (E) Cytolytic activity of control or PRDM1-knockout CAR-T cells against the indicated target cells was analyzed with flow cytometry (n = 3 cultures, unpaired 2-tailed Student t test for each condition). Representative data of 2 experiments. (F-G) Cytolytic activity of control or PRDM1-knockout CAR-T cells against NALM6 (F) or K562-CD19 (G) at the indicated time points (n = 3-4 cultures, one-way ANOVA with multiple comparisons test). (H-L) NSG mice were intravenously infused with NALM6-GL, and then with CAR-T cells with or without PRDM1 knockout 10 days later (n = 10 mice for each CAR). (I) The frequency of human T cells in the PB was analyzed at the indicated time points (unpaired 2-tailed Student t test of the log-transformed values for each time point). (J) Total flux in the whole body was measured by in vivo bioluminescence imaging (unpaired 2-tailed Student t test of the log-transformed values). (K) Kaplan-Meier analysis for leukemia-related survival after NALM6-GL infusion (log-rank test). In (H-K), representative data of 2 independent experiments are shown. (L) The PRDM1-knockout frequency was compared between infusion products and T cells within the spleen of the mice that developed xenogeneic GVHD (n = 4, paired 2-tailed Student t test). (M-N) NALM6-GL-engrafted NSG mice were transplanted with control or PRDM1-knockout CAR-T cells generated in the same protocol as that shown in (H). The frequency of CD8+ or CD4+ CAR-T cells (M) (n = 6, unpaired 2-tailed Student t test of the log-transformed values) or those with TSCM and TCM phenotypes (N) (n = 6, unpaired 2-tailed Student t test for each time point) in the PB was determined by flow cytometry. (O-R) NSG mice subcutaneously inoculated with A375-CD19 (day 0) were treated with CAR-T cells with or without PRDM1 knockout (day 7). The mice were rechallenged with A375-CD19 (day 24) and monitored for tumor progression (n = 9 mice for each group). The data shown are serial monitoring of tumor volume (P), relapse-free survival (Q) (log-rank test), and the frequency of human T cells at the indicated time points (R) (unpaired 2-tailed Student t test of the log-transformed values). In B, E, F, G, I, J, M, N, and R, horizontal lines denote the mean values. NS, not significant.
Previous studies have demonstrated that while Prdm1-knockout mice display enhanced memory T-cell formation, Prdm1−/− T cells exhibit attenuated antigen-induced cytotoxicity.34,35 Consistent with these results, PRDM1 ablation significantly impaired the production of cytolytic molecules, including granzyme B and perforin in CAR-T cells (supplemental Figure 7A-B). PRDM1-knockout CAR-T cells exhibited delayed but potent cytolytic activity against target tumor cells (Figure 3E-G). To evaluate the impact of PRDM1-ablated CAR-T cells on effective antitumor functions in vivo, CAR-T cells with or without PRDM1 knockout were adoptively transferred to NALM6-bearing NSG mice (Figure 3H). PRDM1-knockout CAR-T cells persisted significantly better than the control CAR-T cells (Figure 3I), which resulted in the superior control of leukemia progression (Figure 3J-K; supplemental Figure 8A). Moreover, PRDM1-knockout T cells expanded and induced xenogeneic GVHD in some of the treated mice (supplemental Figure 8B-C; supplemental Table 2), thereby suggesting PRDM1-disrupted CAR-T cells retained their proliferative capacity following the control of leukemia progression. Similar to the results in the tumor-free model, the PRDM1-knockout frequency in the T cells that persisted in the spleen was higher than that of the infusion products (Figure 3L). We further investigated the mechanism by which PRDM1 ablation affects CAR-T cell expansion and differentiation in vivo. On day 7 following T-cell infusion, PRDM1 ablation mitigated CAR-T cell expansion (Figure 3M). However, the persistence of PRDM1-knockout CAR-T cells outperformed the control T cells on days 14 and 24, thus suggesting PRDM1-ablated CAR-T cells displayed a delayed-onset response but eventually better persistence at later time points. Importantly, T cells with an early memory phenotype were substantially maintained in the PRDM1-knockout CAR-T cell population (Figure 3N; supplemental Figure 9A-B), consistent with the in vitro results. The persistence of CD4+ CAR-T cells was also enhanced by PRDM1 knockout despite no prominent difference in the frequency of immature memory T cells at each time point.
Next, we investigated if PRDM1-deficient CAR-T cells exhibited durable antitumor functions in a solid tumor model using the melanoma cell line A375 transduced with CD19 (A375-CD19) (Figure 3O). The infused CAR-T cells efficiently controlled the growth of initially engrafted tumors in all mice (Figure 3P). To evaluate the durability of their therapeutic efficacy, NSG mice were rechallenged with A375-CD19 24 days after the initial transplantation. PRDM1-knockout CAR-T cells demonstrated better control of tumor growth and better persistence than the control CAR-T cells (Figure 3P-R). Taken together, despite PRDM1 knockout attenuating rapid-onset cytolytic activity, it enhanced the in vivo expansion and persistence of CAR-T cells, thereby resulting in improved therapeutic efficacy in both liquid and solid tumor models.
PRDM1 knockout globally alters gene expression and epigenetic profiles of CAR-T cells
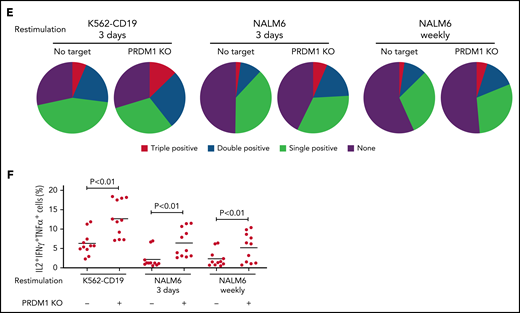
To gain more insight into the mechanism by which PRDM1 deficiency modified CAR-T cell functions, we compared the genome-wide gene expression profiles between PRDM1-knockout and control CD8+ CAR-T cells following repeated antigen stimulation. Overall, 2192 genes were differentially expressed between the groups (false discovery rate [FDR] < 0.05). Unsupervised hierarchical clustering and principal component analysis demonstrated that PRDM1-ablated CAR-T cells formed a distinct cluster from the control CAR-T cells, notwithstanding the variability among different donors (Figure 4A-B). Upregulated genes in PRDM1-knockout CAR-T cells included memory-associated transcription factors and surface molecules (TCF7, LEF1, STAT3, CCR7, and IL7R), while multiple genes induced upon effector differentiation (KLRG1, EOMES, TBX21, and ID2) were significantly downregulated (supplemental Figure 10A-B).42 Some but not all direct or indirect target genes of Blimp1 (ID3, TCF3, MYC, and PAX5) displayed altered expression in PRDM1-knockout T cells (supplemental Figure 10C).32,43 Consistent with the immunoblotting analysis, TOX was significantly upregulated in PRDM1-knockout T cells (supplemental Figure 10D). PRDM1 knockout did not alter the expression of NR4A family genes. The gene set enrichment analysis demonstrated that genes representing the early memory phenotype44-46 were significantly enriched in PRDM1-knockout CAR-T cells, thus corroborating PRDM1 ablation globally remodeled gene expression profiles toward those of less differentiated memory T cells (Figure 4C). We also investigated the epigenetic alteration of CAR-T cells induced by PRDM1 ablation using ATAC sequencing. As observed in the gene expression profiles, PRDM1-knockout CAR-T cells from 2 different donors formed distinct epigenetic signatures compared with the control T cells (Figure 4D-E). The majority of differentially accessible regions were more accessible in PRDM1-knockout CAR-T cells, which was reasonable considering the role of PRDM1 as a transcriptional repressor.47 Consistent with the RNA-sequencing data, PRDM1-knockout CAR-T cells demonstrated increased chromatin accessibility in the promoter regions of memory-related genes, including CCR7 and TCF7 (Figure 4F). We observed a substantial correlation between gene expression and epigenetic changes, thus suggesting PRDM1 ablation induced epigenetic remodeling, which in turn altered the gene expression (Figure 4G). To investigate if a specific PRDM1-regulated gene affected T-cell properties, we focused on transcription factors TCF7 and STAT1, which were robustly upregulated upon PRDM1 knockout at transcriptional, epigenetic, and protein levels (Figure 4H; supplemental Figure 11A-C). While PRDM1 knockout principally upregulated CD62L and CCR7 expression, an ectopic expression of TCF7 in CAR-T cells helped in maintaining other memory markers, such as CD27, CD28, and IL7R (Figure 4I; supplemental Figure 12A). The overexpression of a constitutively active STAT1 mutant slightly improved cytokine polyfunctionality; however, it did not affect memory marker profiles (Figure 4J; supplemental Figure 12B-C). Therefore, the altered transcriptional activity of multiple downstream targets of PRDM1 collectively orchestrated the functional properties of PRDM1-ablated CAR-T cells.

Gene expression and epigenetic architecture of PRDM1-knockout CAR-T cells. (A-F) The CD19-targeting CAR-T cells with or without PRDM1 knockout were repeatedly stimulated with NALM-6 3 times and analyzed for gene expression profiles by RNA-sequencing (A-C) (n = 3 different donors) or epigenetic profiles by ATAC-sequencing (D-F) (n = 2 different donors). The data shown are unsupervised hierarchical clustering (A), principal component analysis (B) of differentially expressed genes (FDR < 0.01), and gene set enrichment analysis between control and PRDM1-knockout CAR-T cells using genes upregulated in T cells with an early memory phenotype (C) (nominal P values are shown). (D) Heatmap of differentially accessible regions between control and PRDM1-ablated CAR-T cells (FDR < 0.05). (E) Log2 fold change in read counts (y-axis) between control and PRDM1-knockout CAR-T cells were plotted against log2 average read counts of all samples (x-axis) for individual peak regions. Red dots denote differentially accessible sites at FDR < 0.05. (F) Chromatin accessibility tracks at the promoter regions of the indicated genes. (G-H) Correlation between RNA-seq and ATAC-seq results in the genes with differential expression and chromatin accessibility at promoter regions (FDR < 0.05). Only transcription factor-encoding genes are shown in H. (I) CAR-T cells with or without ectopic expression of TCF7 or PRDM1 knockout were restimulated with NALM-6 and analyzed for the indicated memory markers 4 days later. Mean fluorescence intensity calculated for each sample was shown (n = 4, one-way ANOVA with multiple comparisons test). (J) CAR-T cells transduced with constitutively active STAT1 (caSTAT1) and/or ablated with PRDM1 were analyzed for cytokine production upon restimulation with NALM-6 (n = 4, one-way ANOVA with multiple comparisons test). In (I-J), horizontal lines denote the mean values. NS, not significant.
Gene expression and epigenetic architecture of PRDM1-knockout CAR-T cells. (A-F) The CD19-targeting CAR-T cells with or without PRDM1 knockout were repeatedly stimulated with NALM-6 3 times and analyzed for gene expression profiles by RNA-sequencing (A-C) (n = 3 different donors) or epigenetic profiles by ATAC-sequencing (D-F) (n = 2 different donors). The data shown are unsupervised hierarchical clustering (A), principal component analysis (B) of differentially expressed genes (FDR < 0.01), and gene set enrichment analysis between control and PRDM1-knockout CAR-T cells using genes upregulated in T cells with an early memory phenotype (C) (nominal P values are shown). (D) Heatmap of differentially accessible regions between control and PRDM1-ablated CAR-T cells (FDR < 0.05). (E) Log2 fold change in read counts (y-axis) between control and PRDM1-knockout CAR-T cells were plotted against log2 average read counts of all samples (x-axis) for individual peak regions. Red dots denote differentially accessible sites at FDR < 0.05. (F) Chromatin accessibility tracks at the promoter regions of the indicated genes. (G-H) Correlation between RNA-seq and ATAC-seq results in the genes with differential expression and chromatin accessibility at promoter regions (FDR < 0.05). Only transcription factor-encoding genes are shown in H. (I) CAR-T cells with or without ectopic expression of TCF7 or PRDM1 knockout were restimulated with NALM-6 and analyzed for the indicated memory markers 4 days later. Mean fluorescence intensity calculated for each sample was shown (n = 4, one-way ANOVA with multiple comparisons test). (J) CAR-T cells transduced with constitutively active STAT1 (caSTAT1) and/or ablated with PRDM1 were analyzed for cytokine production upon restimulation with NALM-6 (n = 4, one-way ANOVA with multiple comparisons test). In (I-J), horizontal lines denote the mean values. NS, not significant.
PRDM1 knockout prevents accelerated differentiation of tonic-signaling CAR-T cells
We examined if PRDM1 knockout provided a therapeutic advantage in antitumor T cells targeting antigens other than CD19. The 28z CAR against GD2 (clone 14g2a with E101K mutation) induces tonic signaling, in which T cells undergo antigen-independent activation.48,49 We confirmed that PRDM1 was efficiently ablated by CRISPR/Cas9 in GD2-targeted CAR-T cells as in CAR-T cells targeting CD19 (Figure 5A). PRDM1 ablation did not prevent tonic signaling-mediated PD1 upregulation in the anti-GD2 28z CAR-T cells (Figure 5B-C). However, the PRDM1-knockout CAR-T cells demonstrated better maintenance of an early memory T-cell phenotype and cytokine polyfunctionality compared with control CAR-T cells (Figure 5D-E; supplemental Figure 13A-C). These tendencies were more prominent in 28z CAR-T cells than those in BBz CAR-T cells. PD1 expression in T cells suppresses IL-2 secretion upon ligation with PDL1.50 However, PRDM1-deficient anti-GD2 CAR-T cells demonstrated increased cytokine production upon stimulation by the PDL1-expressing target cells (supplemental Figure 13D-E). The 28z CAR-T cells further upregulated immunoinhibitory molecules and showed a terminally differentiated phenotype and attenuated cytokine production following repeated antigen stimulation (supplemental Figure 14A-H). The PRDM1-knockout CAR-T cells again revealed an increased frequency of a central memory phenotype and multiple cytokine-producing cells without substantially affecting the upregulation of exhaustion markers.


PRDM1 knockout augments maintenance of a memory phenotype and cytokine polyfunctionality in multiple types of antitumor T cells. (A) Immunoblotting analysis of Blimp-1 expression in control or PRDM1-knockout 28z CAR-T cells targeting GD2. Representative blots of 3 experiments. (B-C) Surface expression of PD1, TIM-3, and LAG3 in CD19 or GD2-targeting 28z or BBz CAR-T cells with or without PRDM1 knockout was analyzed on day 10. Representative flow cytometry plots (B) and mean fluorescence intensity of each molecule (C) (n = 4, one-way ANOVA with multiple comparisons test). Experiments were repeated twice. **P < .01 compared with the control FMC63-28z CAR-T cells. (D-E) The frequency of TSCM/TCM (D) and IL2+IFNγ+TNFα+ cells (E) was analyzed in the CD8+ CAR-T cell population on day 11 (n = 3-4, unpaired 2-tailed Student t test). Representative data of 2 independent experiments. (F-I) NSG mice infused with GD2-expressing NALM6-GL were treated with the 14g2aE101K CAR-T cells with or without PRDM1 knockout (n = 7 for each group). The frequency of human T cells in the PB was analyzed at the indicated time points (G) (unpaired 2-tailed Student t test). Tumor progression was monitored by in vivo bioluminescence imaging (H) (unpaired 2-tailed Student t test of the log-transformed values). Kaplan-Meier curve for the overall survival of mice (I) (n = 7, log-rank test). (J-L) T cells transduced with anti-mesothelin 28z CAR (clone ss1) or HLA-A2/MART127–35-specific TCR (clone DMF5) were ablated with PRDM1 and analyzed for memory markers (J) (n = 4, unpaired 2-tailed Student t test), fold expansion (K) (n = 4, unpaired 2-tailed Student t test), and cytokine production (L) (n = 7 for ss1 CAR-T cells and n = 4 for DMF5 TCR-T cells, unpaired 2-tailed Student t test) after 3 antigenic stimulations. (M) The frequency of PRDM1 knockout was evaluated in the ss1 CAR-T cells before and after stimulation (n = 4 samples). In C-E, G, H, and J-M, horizontal lines depict the mean values. NS, not significant.
PRDM1 knockout augments maintenance of a memory phenotype and cytokine polyfunctionality in multiple types of antitumor T cells. (A) Immunoblotting analysis of Blimp-1 expression in control or PRDM1-knockout 28z CAR-T cells targeting GD2. Representative blots of 3 experiments. (B-C) Surface expression of PD1, TIM-3, and LAG3 in CD19 or GD2-targeting 28z or BBz CAR-T cells with or without PRDM1 knockout was analyzed on day 10. Representative flow cytometry plots (B) and mean fluorescence intensity of each molecule (C) (n = 4, one-way ANOVA with multiple comparisons test). Experiments were repeated twice. **P < .01 compared with the control FMC63-28z CAR-T cells. (D-E) The frequency of TSCM/TCM (D) and IL2+IFNγ+TNFα+ cells (E) was analyzed in the CD8+ CAR-T cell population on day 11 (n = 3-4, unpaired 2-tailed Student t test). Representative data of 2 independent experiments. (F-I) NSG mice infused with GD2-expressing NALM6-GL were treated with the 14g2aE101K CAR-T cells with or without PRDM1 knockout (n = 7 for each group). The frequency of human T cells in the PB was analyzed at the indicated time points (G) (unpaired 2-tailed Student t test). Tumor progression was monitored by in vivo bioluminescence imaging (H) (unpaired 2-tailed Student t test of the log-transformed values). Kaplan-Meier curve for the overall survival of mice (I) (n = 7, log-rank test). (J-L) T cells transduced with anti-mesothelin 28z CAR (clone ss1) or HLA-A2/MART127–35-specific TCR (clone DMF5) were ablated with PRDM1 and analyzed for memory markers (J) (n = 4, unpaired 2-tailed Student t test), fold expansion (K) (n = 4, unpaired 2-tailed Student t test), and cytokine production (L) (n = 7 for ss1 CAR-T cells and n = 4 for DMF5 TCR-T cells, unpaired 2-tailed Student t test) after 3 antigenic stimulations. (M) The frequency of PRDM1 knockout was evaluated in the ss1 CAR-T cells before and after stimulation (n = 4 samples). In C-E, G, H, and J-M, horizontal lines depict the mean values. NS, not significant.
When adoptively transferred to NSG mice harboring GD2-expressing NALM6, PRDM1-knockout CAR-T cells were detected at higher frequencies and delayed tumor progression significantly better than the control CAR-T cells (Figure 5F-I; supplemental Figure 15A-B). PRDM1 ablation in the GD2-targeting BBz CAR-T cells also enhanced T-cell persistence and therapeutic response (supplemental Figure 16A-D). Moreover, we confirmed the effects of PRDM1 ablation on the maintenance of an immature memory phenotype and cytokine polyfunctionality in mesothelin-targeting CAR-T cells and TCR-transduced T cells against HLA-A2 MART1 (Figure 5J-L). The frequency of PRDM1 knockout was maintained after repeated stimulation (Figure 5M). These results suggest that PRDM1 knockout is potentially useful for enhancing the efficacy of CAR- or TCR-engineered T cells that target antigens other than CD19.
PRDM1 knockout partially restores a memory phenotype in terminally differentiated T cells
Finally, we investigated if the ablation of PRDM1 could restore memory T-cell properties in already differentiated T cells. Repeatedly stimulated CAR-T cells were nearly deprived of an early memory phenotype (Figure 6A). Surprisingly, the CAR-T cells regained the expression of some memory markers, such as CCR7, CD62L, and CD28, upon PRDM1 knockout (Figure 6A-B). The T cells ablated with PRDM1 following repeated stimulation displayed significantly better in vivo persistence compared with the control T cells, which hardly revealed long-term persistence (Figure 6C-D).
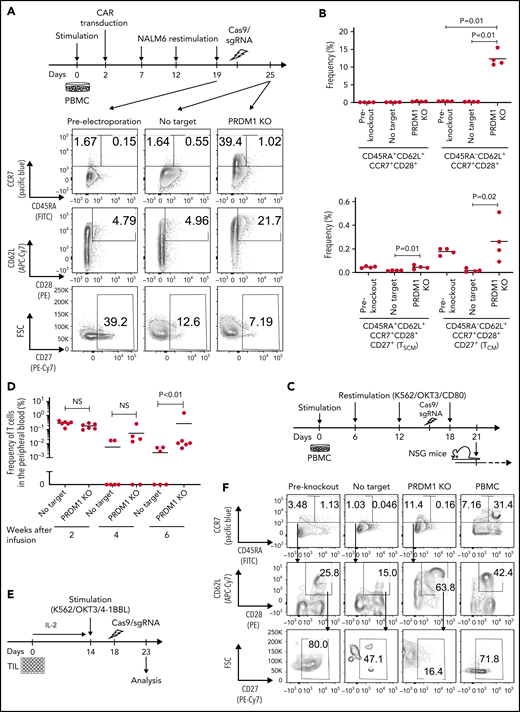

Terminally differentiated T cells partially acquire memory T-cell properties upon PRDM1 knockout. (A-B) CD19-targeting CAR-T cells were electroporated with Cas9/sgRNA against PRDM1 following repeated stimulation by NALM-6 and analyzed for memory T-cell markers. Representative flow cytometry plots (A) and the frequency of the indicated populations are shown (B) (n = 4, ordinary one-way ANOVA with multiple comparisons test). Representative data of 2 experiments. (C,D) Human T cells knocked out with PRDM1 after repeated stimulations were infused into irradiated NSG mice (n = 6 mice per group). The frequency of human T cells in the PB was monitored at the indicated time points (D) (n = 6, Mann-Whitney U test). (E-I) TIL dissociated from tumor samples were expanded and then ablated with PRDM1. Representative flow cytometry plots analyzing memory T-cell phenotypes (F) and the frequency of CD8+ T cells expressing the indicated molecules (G) (n = 8 samples, repeated measures of one-way ANOVA with multiple comparisons test). (H) The expression of TCF7 was analyzed by intracellular flow cytometry 5 days after electroporation. Mean fluorescence intensity within the CD8+ T-cell population was shown (n = 5, paired 2-tailed Student t test). (I) Expanded TILs were restimulated by K562 expressing anti-CD3 mAb and 41BBL, and the production of IL-2, IFN-γ, and TNF-α was analyzed by flow cytometry (n = 5, paired 2-tailed Student t test).
Terminally differentiated T cells partially acquire memory T-cell properties upon PRDM1 knockout. (A-B) CD19-targeting CAR-T cells were electroporated with Cas9/sgRNA against PRDM1 following repeated stimulation by NALM-6 and analyzed for memory T-cell markers. Representative flow cytometry plots (A) and the frequency of the indicated populations are shown (B) (n = 4, ordinary one-way ANOVA with multiple comparisons test). Representative data of 2 experiments. (C,D) Human T cells knocked out with PRDM1 after repeated stimulations were infused into irradiated NSG mice (n = 6 mice per group). The frequency of human T cells in the PB was monitored at the indicated time points (D) (n = 6, Mann-Whitney U test). (E-I) TIL dissociated from tumor samples were expanded and then ablated with PRDM1. Representative flow cytometry plots analyzing memory T-cell phenotypes (F) and the frequency of CD8+ T cells expressing the indicated molecules (G) (n = 8 samples, repeated measures of one-way ANOVA with multiple comparisons test). (H) The expression of TCF7 was analyzed by intracellular flow cytometry 5 days after electroporation. Mean fluorescence intensity within the CD8+ T-cell population was shown (n = 5, paired 2-tailed Student t test). (I) Expanded TILs were restimulated by K562 expressing anti-CD3 mAb and 41BBL, and the production of IL-2, IFN-γ, and TNF-α was analyzed by flow cytometry (n = 5, paired 2-tailed Student t test).
We also determined the effect of PRDM1 knockout on TILs derived from gynecologic or lung cancer (Figure 6E; supplemental Table 3). Most of the expanded TIL samples possessed a terminally differentiated phenotype (Figure 6F).16 Upon PRDM1 knockout, cultured TILs partially restored CD62L and CCR7 expression, as observed in repeatedly stimulated CAR-T cells (Figure 6G). PRDM1-disrupted TILs also displayed increased TCF7 expression and polyfunctional cytokine production upon restimulation (Figure 6H-I). These results collectively demonstrated that differentiated T cells could partially restore early memory T-cell phenotypes and functions by the genetic knockout of PRDM1.
Discussion
The balance between memory and effector T cell functions is required for the T cells infused in vivo to induce potent and durable therapeutic efficacy. PRDM1-disrupted CAR-T cells acquire the potential of greater survival equipped with memory T cells and demonstrate impaired cytotoxic functions, which could negatively affect the antitumor response. Our findings indicated PRDM1 knockout as a suitable approach, at least for CD19-directed CAR-T cells, to induce a sustained antitumor response. Considering the less prominent therapeutic advantage of PRDM1-ablated CAR-T cells targeting GD2, the feasibility of this strategy may depend on multiple factors, such as target antigens and the avidity of antitumor T cells.
While PRDM1-knockout T cells displayed increased cytokine production and superior proliferation in vivo, they revealed increased expression of TOX, which regulates T-cell exhaustion. Previous mouse studies have demonstrated that Tox-deleted T cells acquire gene expression and epigenetic profiles associated with terminal effector differentiation and instead lose the memory T-cell gene signature.36 Tox-knockout T cells initially display potent effector functions; however, Tox-knockout antitumor or antiviral T cells were inferior in long-term survival because of accelerated effector differentiation.37,38,51 Increased TOX expression in PRDM1-knockout T cells may contribute to the acquisition of long-term survival capacity by suppressing the over-activation of antitumor T cells. Alternatively, the inhibition of TOX or other exhaustion-associated factors, such as NR4A and AP-1 transcription factors, may further enhance the antitumor response of PRDM1-knockout T cells by downregulating inhibitory receptors.36,49,52
PRDM1 knockout did not affect the expression of some canonical Blimp1-regulated genes.43 The effect of PRDM1 knockout may vary depending on the T cell conditions (eg, naïve, memory, and effector). Transient suppression of PRDM1 at different time points will provide further insights into the role of Blimp-1 in T cells. Moreover, the effect of PRDM1 knockout on maintaining an early memory phenotype was weaker in CD4+ T cells than in CD8+ T cells. It would be necessary to elucidate how PRDM1 ablation modulates CD4+ T-cell functions at epigenetic and gene expression levels.
PRDM1 knockout not only supported the maintenance of an early memory phenotype but also restored a portion of memory T-cell profiles in terminally differentiated T cells. The direct dedifferentiation of terminally differentiated T cells into early memory T cells will initiate a revolutionary improvement in TIL therapy, in which extracted antitumor T cells have often acquired a terminally differentiated phenotype.53 PRDM1 knockout in TIL samples did not restore the expression of several memory markers, such as CD27 and CD28. This warrants additional modification to completely reprogram terminally differentiated T cells into early memory T cells.
In summary, the present study provided a rationale that PRDM1 knockout in antitumor T cells enabled long-term persistence and durable therapeutic response through the epigenetic modulation of T-cell differentiation programs. These findings are potentially applicable to a wide range of adoptive cancer immunotherapies.
Acknowledgments
The authors thank Editage (http://www.editage.com) for editing and reviewing the English in the manuscript.
This work was supported by AMED under Grant Number JP20ae0201013 and JP21bm0704066 (Y.K.), JSPS KAKENHI Grant Number 20H03543 and 19K22552 (Y.K.), Takara Bio, Inc (Y.K.), Aichi Cancer Center Joint Research Project on Priority Areas (Y.K. and H.M.), the Ichiro Kanehara Foundation (Y.K.), the Yasuda Medical Foundation (Y.K.), the Senri Life Science Foundation (Y.K.), the Princess Takamatsunomiya Cancer Research Foundation (Y.K.), Takeda Science Foundation (Y.K.), Senshin Medical Research Foundation (Y.K.), Uehara Memorial Foundation (Y.K.), Japan Leukemia Research Fund (Y.K.), Japanese Society of Hematology Research Grant (Y.K.), Astellas Foundation for Research on Metabolic Disorders (Y.K.), Mochida Memorial Foundation for Medical and Pharmaceutical Research (Y.K.), SGH Foundation (Y.K.), Tokyo Biochemical Research Foundation (Y.K.), Nippon Shinyaku Research Grant (Y.K.), Bristol-Myers Squibb Research Grant (Y.K.), KAKENHI Grant Number 19K09297 (T.Y.), KAKENHI Grant Number 20K22793 (Z.W.), and KAKENHI Grant Number 19H03528 (H.M.).
Authorship
Contribution: Y.K. designed the project; T.Y., Z.W., S.I., H. Kasuya, Y.T., and Y.K. performed the experiments; H.M., H. Kuroda, W.H., and S.S. provided critical human TIL samples and contributed to the writing of this manuscript; and T.Y. and Y.K. analyzed the results and wrote the manuscript.
Conflict-of-interest disclosure: This study was partly supported by a commercial research grant from Takara Bio, Inc. to Y.K.
Correspondence: Yuki Kagoya, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan; e-mail: y.kagoya@aichi-cc.jp.
The RNA-seq and ATAC-seq datasets presented in Figure 4 have been deposited in the NCBI Gene Expression Omnibus (accession numbers GSE173918 and GSE173620).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal