

Key Points
CD19 CAR T-cell therapy is effective at achieving durable remission for relapsed/refractory ALL across cytogenetic risk groups.
CD19 CAR T-cell treatment results for patients with high-risk cytogenetics including Ph+, Ph-like, and KMT2A-rearranged ALL are encouraging.

Abstract
Chimeric antigen receptor (CAR) T-cell therapy can induce durable remissions of relapsed/refractory B-acute lymphoblastic leukemia (ALL). However, case reports suggested differential outcomes mediated by leukemia cytogenetics. We identified children and young adults with relapsed/refractory CD19+ ALL/lymphoblastic lymphoma treated on 5 CD19-directed CAR T-cell (CTL019 or humanized CART19) clinical trials or with commercial tisagenlecleucel from April 2012 to April 2019. Patients were hierarchically categorized according to leukemia cytogenetics: High-risk lesions were defined as KMT2A (MLL) rearrangements, Philadelphia chromosome (Ph+), Ph-like, hypodiploidy, or TCF3/HLF; favorable as hyperdiploidy or ETV6/RUNX1; and intermediate as iAMP21, IKZF1 deletion, or TCF3/PBX1. Of 231 patients aged 1 to 29, 74 (32%) were categorized as high risk, 28 (12%) as intermediate, 43 (19%) as favorable, and 86 (37%) as uninformative. Overall complete remission rate was 94%, with no difference between strata. There was no difference in relapse-free survival (RFS; P = .8112), with 2-year RFS for the high-risk group of 63% (95% confidence interval [CI], 52-77). There was similarly no difference seen in overall survival (OS) (P = .5488), with 2-year OS for the high-risk group of 70% (95% CI, 60-82). For patients with KMT2A-rearranged infant ALL (n = 13), 2-year RFS was 67% (95% CI, 45-99), and OS was 62% (95% CI, 40-95), with multivariable analysis demonstrating no increased risk of relapse (hazard ratio, 0.70; 95% CI, 0.21-2.90; P = .7040) but a higher proportion of relapses associated with myeloid lineage switch and a 3.6-fold increased risk of all-cause death (95% CI, 1.04-12.75; P = .0434). CTL019/huCART19/tisagenlecleucel are effective at achieving durable remissions across cytogenetic categories. Relapsed/refractory patients with high-risk cytogenetics, including KMT2A-rearranged infant ALL, demonstrated high RFS and OS probabilities at 2 years.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy has radically improved the outcomes for children and young adults with relapsed/refractory B-acute lymphoblastic leukemia (ALL), with 1-year relapse-free survival (RFS) rates approaching 60%.1-5 Although leukemic genomic abnormalities are prognostic biomarkers of outcome at initial diagnosis, with risk associations preserved at relapse,6 their clinical implication in novel therapeutic approaches such as CAR T-cell therapy are unknown. As specific genetic aberrations are known to influence overall response to cytotoxic therapy and to specific chemotherapy agents,7 and initial case series in CAR T-cell therapy suggested differential results mediated by certain cytogenetic characteristics,8 although other preliminary data contradicted those findings,9,10 understanding the clinical outcomes of patients with common cytogenetic and genomic aberrations treated with CAR T cells is essential.
Cytogenetic and genomic abnormalities that are considered to confer a poor prognosis include KMT2A (formerly MLL) rearrangements, the Philadelphia chromosome (Ph+), fusions and mutations that confer a Philadelphia chromosome–like (Ph-like) gene expression profile, hypodiploidy, and TCF3/HLF fusion resulting from t(17;19)(q22;p13). Limited prospective clinical trial data using CD19-directed CAR T-cell constructs suggests that there is no initial treatment response difference seen with Ph+ ALL11 or those with KMT2A rearrangements.8 However, CAR T-cell escape variants, including myeloid lineage switch, have been identified, most commonly with KMT2A rearrangement8 but also with TCF3-ZNF384 fusions12 and in those without defined high-risk cytogenetic features.13,14
The presence of hyperdiploidy or ETV6/RUNX1 fusion at initial diagnosis typically connotes a favorable prognosis,15-18 although not all cases fare evenly,19,20 and there has not been explicit examination of these features in the context of CAR T-cell therapy. Similarly, there is no available data regarding intrachromosomal amplification of chromosome 21 (iAMP21) or TCF3/PBX1 fusion resulting from t(1;19)(q23;p13.3) in the setting of CAR T cells, with both lesions classified as intermediate risk using intensive chemotherapy in contemporary protocols.21-24IKZF1 deletions, often associated with Ph+ and Ph-like,25,26 carry a poor prognosis both at diagnosis27 and at relapse,28 which is likely impacted by multiple other factors29 and likewise have not been explored in the context of CAR T-cell therapy.
To examine whether the efficacy of CD19-directed CAR T-cell therapy for B-ALL differs based on sentinel cytogenetic lesions, we conducted a retrospective outcomes analysis of children and young adults treated with murine CTL019/tisagenlecleucel or the related humanized CD19 CAR, huCART19,30 for relapsed/refractory ALL or lymphoblastic lymphoma (LLy) categorized by cytogenetic risk level.
Methods
Study population
Children and young adults aged 1 to 29 years with relapsed or refractory CD19+ ALL or LLy treated on 5 CD19-directed CAR T-cell clinical trials (clinicaltrial.gov numbers NCT01626495, NCT02435849, NCT02374333, NCT02228096, and NCT02906371)1,2,30-32 or with commercial tisagenlecleucel (Kymriah, Novartis) at the Children’s Hospital of Philadelphia from April 2012 to April 2019 were included (CONSORT diagram, supplemental Figure 1). Most patients received CTL019/tisagenlecleucel (n = 195), an anti-CD19 CAR containing a 4-1BB costimulatory domain.33 Patients on trial NCT02374333 (n = 41) received huCART19, in which the anti-CD19 scFv has been humanized.30 Patients who were treated in the retreatment cohort of NCT02374333 were not included in these analyses. Patients who lacked cytogenetic analysis of their leukemia were excluded (n = 5). This retrospective study was approved by the institutional review board of the Children’s Hospital of Philadelphia. Patients or their guardians provided written informed consent for treatment in each respective clinical trial.
Prior to infusion, lymphodepleting (LD) chemotherapy was administered at the treating physician’s discretion. The recommended agents were fludarabine (30 mg/m2 per day × 4 days) and cyclophosphamide (500 mg/m2 per day × 2 days) (n = 213, alternative regimens presented in supplemental Table 1). Patients underwent staging bone marrow aspirate and biopsy with multiparameter flow cytometry determination of minimal residual disease (MRD) after lymphodepleting chemotherapy and prior to infusion, with the exception of 23 patients who underwent staging disease evaluation at enrollment and received bridging chemotherapy prior to infusion. Preinfusion disease burden was defined based on the highest blast percentage of the 3 measurements: M1, <5% lymphoblasts; M2, 5% to 25% lymphoblasts; and M3, >25% lymphoblasts.
Cytogenetic risk group definitions
Cytogenetic information was abstracted from the medical record, clinical trial database, and referral documentation. Patients were classified according to their highest risk cytogenetic characteristic and stratified by cytogenetic risk category accordingly. In the case of multiple cytogenetic reports, the highest risk cytogenetic characteristic documented was used in stratification, relying on known preservation of sentinel lesions in relapse.6,34-36 High-risk lesions were defined as KMT2A (MLL) rearrangements, Philadelphia chromosome (Ph+), Ph-like,37 hypodiploidy (<44 chromosomes), and TCF3/HLF fusion. Leukemias classified as Ph-like were identified either by gene expression profiling, including low density array screen,38 or by targeted sequencing or fluorescence in situ hybridization for specific lesions. Intermediate risk lesions included iAMP21, IKZF1 deletion, or TCF3/PBX1. Favorable cytogenetics were defined as the presence of hyperdiploidy (>51 chromosomes) or ETV6/RUNX1 fusion. If none of the preceding lesions were identified, the leukemia was classified as having uninformative cytogenetics. For patients with multiple coexisting lesions, the highest-risk cytogenetic characteristic was used for risk classification; for example, a patient with Ph+ leukemia that also exhibited an IKZF1 deletion was categorized as high risk.
Outcomes
Complete remission (CR) was defined as bone marrow with trilineage hematopoiesis and <5% lymphoblasts and no evidence of extramedullary leukemia. Event-free survival (EFS) was defined as time from CAR T-cell infusion until evidence of nonresponse, morphologic relapse, or death. RFS was defined as the time from first disease assessment post–CAR T-cell infusion until morphologic relapse or death for those patients who achieved CR following infusion. No death occurred before relapse in these patients. Patients were censored at the time of alternate cancer-directed therapy (including tyrosine kinase inhibitor and hematopoietic stem cell transplantation [HSCT]) or at last contact, whichever was earlier for both RFS and EFS analyses. Patients were not censored for CAR T-cell reinfusion. A secondary RFS analysis, using flow MRD >0.01% as the relapse endpoint, was also performed for individual high-risk lesions vs all others. Overall survival (OS) was defined as time to all-cause death, with patients censored at the last known contact. In addition to clinically-indicated testing, clinical trial participants had routine disease assessments, including lumbar puncture, bone marrow aspirate, and biopsy at months 3, 6, 9, and 12, and patients who received commercial tisagenlecleucel had the same assessments performed at month 3 following CAR T-cell infusion.
Covariates
Patient demographics and baseline characteristics were obtained from the clinical trial databases for trial patients and from the medical record for patients receiving commercial tisagenlecleucel. Patient clinical history, including prior treatment, and disease status at referral were manually abstracted from the medical record.
Statistical analysis
Data used in these analyses were current as of 31 December 2019. Baseline patient and disease characteristics were summarized and compared by cytogenetic risk group using Fisher’s exact tests for categorical variables and Wilcoxon test for continuous variables. Kaplan-Meier curves of RFS and OS were plotted by cytogenetic risk group and compared using log-rank tests. Cumulative incidence function of relapse was also estimated considering alternate cancer-directed therapy as a competing risk (nonrelapse death was not considered as a competing risk because no death occurred before relapse in this study). Cumulative incidence function of relapse was plotted by cytogenetic risk group and compared using Gray’s test. Univariate and multivariate Cox regressions were used to estimate the unadjusted and adjusted hazard ratios (HRs) of RFS and OS for cytogenetic groups, with proportional hazard assumptions assessed by log-log plots. All the baseline characteristics were screened as potential confounders by evaluating each covariate’s bivariate association with the exposure (cytogenetic group) and the outcome (RFS or OS). The covariates that demonstrated some evidence of association (P < .2) with both the exposure and the outcome were then included in the multivariate regression model to control for confounders. Analyses were performed using SAS 9.4 (SAS Institute, Cary, NC) statistical software.
Results
Study population
From April 2012 to April 2019, 236 patients (median age 12 years, range 1-29) with relapsed or refractory CD19+ ALL or LLy were treated on 5 CD19-directed CAR T-cell clinical trials, or received tisagenlecleucel, of which 231 patients had documentation of the cytogenetic characteristics of their disease (supplemental Figure 1). Of the 231, 74 (32%) met criteria to be included in the high-risk cytogenetic strata: 25 (34%) patients with KMT2A rearrangements (fusion partners in supplemental Table 2), 18 (24%) Ph+, 19 (26%) Ph-like, 8 (11%) hypodiploid, and 4 (5.4%) with TCF3/HLF. Twenty-eight (12%) patients met criteria for intermediate risk: 10 (36%) patients with iAMP21, 10 (36%) with IKZF1 deletions, and 8 (29%) with TCF3/PBX1. Forty-three patients (19%) had favorable risk cytogenetics, with 30 (70%) demonstrating hyperdiploidy and 13 (30%) with ETV6/RUNX1. Eighty-six patients (37%) did not have any of the preceding lesions and were therefore categorized in the uninformative risk stratum. Demographic differences in patient sex and age were seen across the strata (P = .029 and P = .047, respectively), whereas distributions of clinical characteristics, such as prior HSCT, number of prior relapses at referral, and bone marrow disease burden at infusion, were comparable (Table 1; supplemental Table 3). Fifteen patients (6.5%) were initially diagnosed at <1 year of age, of whom 13 had KMT2A rearrangements (referred to subsequently as KMT2A-rearranged infant ALL). Ten percent of the cohort received prior treatment with blinatumomab, more commonly in the high-risk and intermediate-risk strata than in the favorable-risk or uninformative categories (P = .040, Table 1).
Baseline patient and disease characteristics
| All patients (n = 231) | High risk (n = 74) | Intermediate risk (n = 28) | Favorable risk (n = 43) | Uninformative (n = 86) | P | |
|---|---|---|---|---|---|---|
| Age (y), median (range) | 12 (1-29) | 11 (1-29) | 9 (2-22) | 12 (4-29) | 13 (2-27) | .047 |
| Age categories, n (%) | ||||||
| <3 y | 12 (5.2%) | 8 (11%) | 2 (7.1%) | 0 (0%) | 2 (2.3%) | .015 |
| 3-9.99 y | 81 (35%) | 23 (31%) | 15 (54%) | 14 (33%) | 29 (34%) | |
| 10-17.99 y | 100 (43%) | 29 (39%) | 10 (36%) | 25 (58%) | 36 (42%) | |
| ≥18 y | 38 (16%) | 14 (19%) | 1 (3.6%) | 4 (9.3%) | 19 (22%) | |
| Male n (%) | 134 (58%) | 49 (66%) | 10 (36%) | 28 (65%) | 47 (55%) | .029 |
| Prior HSCT | 100 (43%) | 38 (51%) | 12 (43%) | 14 (33%) | 36 (42%) | .26 |
| Prior blinatumomab | 24 (10%) | 9 (12%) | 7 (25%) | 2 (4.7%) | 6 (7.0%) | .040 |
| Refractory at referral | 91 (39%) | 30 (41%) | 10 (36%) | 17 (40%) | 34 (40%) | .98 |
| Disease status at referral | ||||||
| Primary refractory | 39 (17%) | 18 (24%) | 2 (7.1%) | 3 (7.0%) | 16 (19%) | .077 |
| 1st relapse | 69 (36%) | 20 (27%) | 12 (43%) | 10 (23%) | 27 (31%) | |
| 2nd or greater relapse | 123 (64%) | 36 (49%) | 14 (50%) | 30 (70%) | 43 (50%) | |
| Marrow status preinfusion | ||||||
| <0.01% | 87 (38%) | 34 (46%) | 7 (25%) | 17 (40%) | 29 (34%) | .18 |
| 0.01-4.99% | 46 (20%) | 16 (22%) | 3 (11%) | 6 (14%) | 21 (24%) | |
| 5-24.99% | 21 (9.1%) | 4 (5.4%) | 3 (11%) | 6 (14%) | 8 (9.3%) | |
| ≥25% | 77 (33%) | 20 (27%) | 15 (54%) | 14 (33%) | 28 (33%) |
| All patients (n = 231) | High risk (n = 74) | Intermediate risk (n = 28) | Favorable risk (n = 43) | Uninformative (n = 86) | P | |
|---|---|---|---|---|---|---|
| Age (y), median (range) | 12 (1-29) | 11 (1-29) | 9 (2-22) | 12 (4-29) | 13 (2-27) | .047 |
| Age categories, n (%) | ||||||
| <3 y | 12 (5.2%) | 8 (11%) | 2 (7.1%) | 0 (0%) | 2 (2.3%) | .015 |
| 3-9.99 y | 81 (35%) | 23 (31%) | 15 (54%) | 14 (33%) | 29 (34%) | |
| 10-17.99 y | 100 (43%) | 29 (39%) | 10 (36%) | 25 (58%) | 36 (42%) | |
| ≥18 y | 38 (16%) | 14 (19%) | 1 (3.6%) | 4 (9.3%) | 19 (22%) | |
| Male n (%) | 134 (58%) | 49 (66%) | 10 (36%) | 28 (65%) | 47 (55%) | .029 |
| Prior HSCT | 100 (43%) | 38 (51%) | 12 (43%) | 14 (33%) | 36 (42%) | .26 |
| Prior blinatumomab | 24 (10%) | 9 (12%) | 7 (25%) | 2 (4.7%) | 6 (7.0%) | .040 |
| Refractory at referral | 91 (39%) | 30 (41%) | 10 (36%) | 17 (40%) | 34 (40%) | .98 |
| Disease status at referral | ||||||
| Primary refractory | 39 (17%) | 18 (24%) | 2 (7.1%) | 3 (7.0%) | 16 (19%) | .077 |
| 1st relapse | 69 (36%) | 20 (27%) | 12 (43%) | 10 (23%) | 27 (31%) | |
| 2nd or greater relapse | 123 (64%) | 36 (49%) | 14 (50%) | 30 (70%) | 43 (50%) | |
| Marrow status preinfusion | ||||||
| <0.01% | 87 (38%) | 34 (46%) | 7 (25%) | 17 (40%) | 29 (34%) | .18 |
| 0.01-4.99% | 46 (20%) | 16 (22%) | 3 (11%) | 6 (14%) | 21 (24%) | |
| 5-24.99% | 21 (9.1%) | 4 (5.4%) | 3 (11%) | 6 (14%) | 8 (9.3%) | |
| ≥25% | 77 (33%) | 20 (27%) | 15 (54%) | 14 (33%) | 28 (33%) |
Outcomes
Disease response and relapse
Overall CR rate was 94%, with no statistically significant difference in CR rate seen between the strata, with 93% (69/74) of high-risk stratum, 86% (24/28) of intermediate risk, 98% (42/43) of favorable risk, and 97% (83/86) of patients with uninformative cytogenetics in CR at day 28 after infusion (P = .23, Table 2).
Postinfusion outcomes by cytogenetic group
| High risk | Intermediate risk | Favorable risk | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MLL (KMT2Ar) (n = 25) | Ph+ (n = 18) | Ph-like (n = 19) | Hypodiploid (n = 8) | TCF3/HLF (n = 4) | iAMP21 (n = 10) | IZF1 (n = 10) | TCF3/PBX1 (n = 8) | Hyperdiploid (n = 30) | ETV6/RUNX1 (n = 13) | Uninformative (n = 86) | |
| Disease response | |||||||||||
| Complete response | 23 (92%) | 18 (100%) | 16 (84%) | 8 (100%) | 4 (100%) | 10 (100%) | 8 (80%) | 6 (75%) | 29 (97%) | 13 (100%) | 83 (97%) |
| No response | 1 (4.0%) | 0 (0%) | 2 (11%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (10%) | 2 (25%) | 1 (3.3%) | 0 (0%) | 3 (3.5%) |
| Inevaluable* | 1 (4.0%) | 0 (0%) | 1 (5.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (10%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Patients with relapse, n (%) | 12 (48%) | 2 (11%) | 3 (16%) | 5 (62%) | 2 (50%) | 2 (20%) | 2 (20%) | 4 (50%) | 6 (20%) | 8 (62%) | 32 (37%) |
| CD19− | 7† | 2 | 2 | 3 | 1 | 0 | 2 | 2 | 3 | 4 | 16‡ |
| CD19+ | 4 | 0 | 0 | 1 | 1 | 2 | 0 | 2 | 2 | 4 | 14 |
| Months of follow-up, median (interquartile range) | 26 (24, 43) | 24 (21, 60) | 30 (15, 45) | 28 (20, 43) | |||||||
| High risk | Intermediate risk | Favorable risk | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MLL (KMT2Ar) (n = 25) | Ph+ (n = 18) | Ph-like (n = 19) | Hypodiploid (n = 8) | TCF3/HLF (n = 4) | iAMP21 (n = 10) | IZF1 (n = 10) | TCF3/PBX1 (n = 8) | Hyperdiploid (n = 30) | ETV6/RUNX1 (n = 13) | Uninformative (n = 86) | |
| Disease response | |||||||||||
| Complete response | 23 (92%) | 18 (100%) | 16 (84%) | 8 (100%) | 4 (100%) | 10 (100%) | 8 (80%) | 6 (75%) | 29 (97%) | 13 (100%) | 83 (97%) |
| No response | 1 (4.0%) | 0 (0%) | 2 (11%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (10%) | 2 (25%) | 1 (3.3%) | 0 (0%) | 3 (3.5%) |
| Inevaluable* | 1 (4.0%) | 0 (0%) | 1 (5.3%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (10%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Patients with relapse, n (%) | 12 (48%) | 2 (11%) | 3 (16%) | 5 (62%) | 2 (50%) | 2 (20%) | 2 (20%) | 4 (50%) | 6 (20%) | 8 (62%) | 32 (37%) |
| CD19− | 7† | 2 | 2 | 3 | 1 | 0 | 2 | 2 | 3 | 4 | 16‡ |
| CD19+ | 4 | 0 | 0 | 1 | 1 | 2 | 0 | 2 | 2 | 4 | 14 |
| Months of follow-up, median (interquartile range) | 26 (24, 43) | 24 (21, 60) | 30 (15, 45) | 28 (20, 43) | |||||||
Three patients died prior to day 28 evaluation from sequelae of cytokine release syndrome, coagulopathy, and infection without clear evidence of progressive disease.
Includes 4 patients with lineage switch.
Includes 1 patient with lineage switch.
Median length of follow-up was 27 months and comparable across strata (P > .9, Table 2), with no difference in EFS (P = .7755; 2-year EFS: 59% [95% CI, 48-73] high risk, 50% [95% CI, 33-76] intermediate, 61% [95% CI, 45-83] favorable, and 56% [95% CI, 45-70] uninformative; supplemental Figure 2). RFS also did not differ between the 4 strata (P = .8112; 2-year RFS: 63% [95% CI, 52-77] high risk, 59% [95% CI ,40-86] intermediate, 63% [95% CI, 47-84] favorable, and 55% [95% CI, 43-70] uninformative; Figure 1A). Likewise, cumulative incidence of relapse did not differ across strata, with a 2-year estimate of 33% [95% CI, 22-44] for high-risk lesions, 35% [95% CI, 16-55] for intermediate, 29% [95% CI, 16-45] for favorable, and 37% [95% CI, 26-48] for uninformative (P = .8112, supplemental Figure 3).
In a separate analysis of patients with the highest frequency high-risk lesions, there was no difference in RFS for KMT2A rearrangements (P = .1326, Figure 1B) or Ph-like disease (P = .4037, Figure 1D), with 2-year RFS of 46% (95% CI, 29-73) for those with KMT2A rearrangements compared with 62% (95% CI, 54-71) for all others and 92% (95% CI, 79-100) for those with Ph-like disease compared with 58% (95% CI, 51-67) for all others. RFS was improved for those with Ph+ALL (P = .0211, Figure 1C), with 2-year RFS of 88% (95% CI, 74-100) compared with 57% (95% CI, 49-66) for all others. A subgroup analysis for patients with favorable lesions demonstrated no difference in RFS for hyperdiploid (P = .0769) or ETV6/RUNX1 (P = .1899, supplemental Figure 4), although the former trended toward statistical significance. Two-year RFS for hyperdiploid was 64% (95% CI, 42-96) compared with 59% (95% CI, 51-67) for all others, and 2-year RFS for ETV6/RUNX1 was 53% (95% CI, 33-89) compared with 60% (95% CI, 53-69) for all others. Similarly, no difference was seen in the frequency of CD19 positivity at relapse between the strata, with approximately half (42/78, 54%) of relapses being CD19− (P = .89). In multivariate analysis, adjusting for age, sex, blinatumomab exposure, and bone marrow disease burden at infusion, no risk category emerged as associated with a higher hazard of relapse (Table 3). Multivariable analyses examining individual genomic lesions (KMT2A-rearrangment, Ph+, Ph-like, hyperdiploid, ETV6/RUNX1) compared with all others, adjusted for age, sex, blinatumomab exposure, and bone marrow disease at infusion, found no significant risk associations with relapse, although there was a nonsignificant trend toward increased risk for KMT2A rearrangement and similar nonsignificant trend toward decreased risk for hyperdiploid (KMT2A: adjusted HR, 1.94 [95% CI, 0.90-4.18], P = .0892; Ph+: adjusted HR, 0.30 [95% CI, 0.07-1.27], P = .1021; Ph-like: adjusted HR, 0.69 [95% CI, 0.21-2.27], P = .5366; hyperdiploid: adjusted HR, 0.45 [95% CI, 0.18-1.12], P = .0855; ETV6/RUNX1: adjusted HR, 1.5 [95% CI, 0.71-3.29], P = .2797; Table 4).
Univariate and multivariate Cox regression model for RFS and OS by cytogenetic group
| RFS | OS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | |
| Cytogenetics group Favorable risk High risk Intermediate risk Uninformative | REF 1.179 1.045 1.335 | REF 0.600-2.317 0.433-2.525 0.701-2.545 | REF .6320 .9215 .3796 | REF 1.564 0.693 1.489 | REF 0.745-3.284 0.272-1.765 0.757-2.927 | REF .2374 .4416 .2485 | REF 1.373 1.774 1.187 | REF 0.669-2.817 0.769-4.095 0.584-2.413 | REF .3879 .1792 .6352 | REF 1.728 1.059 1.317 | REF 0.799-3.736 0.439-2.553 0.633-2.742 | REF .1643 .8984 .4611 |
| RFS | OS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | |
| Cytogenetics group Favorable risk High risk Intermediate risk Uninformative | REF 1.179 1.045 1.335 | REF 0.600-2.317 0.433-2.525 0.701-2.545 | REF .6320 .9215 .3796 | REF 1.564 0.693 1.489 | REF 0.745-3.284 0.272-1.765 0.757-2.927 | REF .2374 .4416 .2485 | REF 1.373 1.774 1.187 | REF 0.669-2.817 0.769-4.095 0.584-2.413 | REF .3879 .1792 .6352 | REF 1.728 1.059 1.317 | REF 0.799-3.736 0.439-2.553 0.633-2.742 | REF .1643 .8984 .4611 |
Multivariate model adjusted for age, sex, blinatumomab exposure, and bone marrow disease at infusion.
Univariate and multivariate Cox regression model for RFS and OS by individual cytogenetic lesion
| RFS | OS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | |
| KMT2A rearrangement, all | ||||||||||||
| All others KMT2Ar, all | REF 1.597 | REF 0.862-2.958 | REF .1365 | REF 1.943 | REF 0.903-4.179 | REF .0892 | REF 1.899 | REF 0.996-3.619 | REF .0514 | REF 2.589 | REF 1.153-5.811 | REF .0212 |
| KMT2A rearrangement, infant | ||||||||||||
| All others KMT2Ar, infant leukemia* | REF 0.966 | REF 0.353-2.649 | REF .9471 | REF 0.774 | REF 0.207-2.897 | REF .7040 | REF 1.804 | REF 0.724-4.498 | REF .2052 | REF 3.639 | REF 1.039-12.746 | REF .0434 |
| Ph+ | ||||||||||||
| All others Ph+ | REF 0.222 | REF 0.054-0.904 | REF .0356 | REF 0.304 | REF 0.073-1.267 | REF .1021 | REF 0.305 | REF 0.075-1.245 | REF .0981 | REF 0.409 | REF 0.098-1.713 | REF .2211 |
| Ph-like | ||||||||||||
| All others Ph-like | REF 0.614 | REF 0.193-1.952 | REF .4087 | REF 0.686 | REF 0.208-2.267 | REF .5366 | REF 0.701 | REF 0.255-1.924 | REF .4903 | REF 0.777 | REF 0.274-2.203 | REF .6351 |
| Hyperdiploid | ||||||||||||
| All others Hyperdiploid | REF 0.451 | REF 0.182-1.117 | REF .0851 | REF 0.445 | REF 0.177-1.120 | REF .0855 | REF 0.577 | REF 0.250-1.333 | REF .1984 | REF 0.572 | REF 0.243-1.345 | REF .2003 |
| ETV6/RUNX1 | ||||||||||||
| All others ETV6/RUNX1 | REF 1.626 | REF 0.780-3.388 | REF .1946 | REF 1.527 | REF 0.709-3.290 | REF .2797 | REF 1.201 | REF 0.483-2.984 | REF .6936 | REF 1.209 | REF 0.475-3.077 | REF .6903 |
| RFS | OS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | Unadjusted HR | Unadjusted 95% CI | Unadjusted P | Adjusted HR | Adjusted 95% CI | Adjusted P | |
| KMT2A rearrangement, all | ||||||||||||
| All others KMT2Ar, all | REF 1.597 | REF 0.862-2.958 | REF .1365 | REF 1.943 | REF 0.903-4.179 | REF .0892 | REF 1.899 | REF 0.996-3.619 | REF .0514 | REF 2.589 | REF 1.153-5.811 | REF .0212 |
| KMT2A rearrangement, infant | ||||||||||||
| All others KMT2Ar, infant leukemia* | REF 0.966 | REF 0.353-2.649 | REF .9471 | REF 0.774 | REF 0.207-2.897 | REF .7040 | REF 1.804 | REF 0.724-4.498 | REF .2052 | REF 3.639 | REF 1.039-12.746 | REF .0434 |
| Ph+ | ||||||||||||
| All others Ph+ | REF 0.222 | REF 0.054-0.904 | REF .0356 | REF 0.304 | REF 0.073-1.267 | REF .1021 | REF 0.305 | REF 0.075-1.245 | REF .0981 | REF 0.409 | REF 0.098-1.713 | REF .2211 |
| Ph-like | ||||||||||||
| All others Ph-like | REF 0.614 | REF 0.193-1.952 | REF .4087 | REF 0.686 | REF 0.208-2.267 | REF .5366 | REF 0.701 | REF 0.255-1.924 | REF .4903 | REF 0.777 | REF 0.274-2.203 | REF .6351 |
| Hyperdiploid | ||||||||||||
| All others Hyperdiploid | REF 0.451 | REF 0.182-1.117 | REF .0851 | REF 0.445 | REF 0.177-1.120 | REF .0855 | REF 0.577 | REF 0.250-1.333 | REF .1984 | REF 0.572 | REF 0.243-1.345 | REF .2003 |
| ETV6/RUNX1 | ||||||||||||
| All others ETV6/RUNX1 | REF 1.626 | REF 0.780-3.388 | REF .1946 | REF 1.527 | REF 0.709-3.290 | REF .2797 | REF 1.201 | REF 0.483-2.984 | REF .6936 | REF 1.209 | REF 0.475-3.077 | REF .6903 |
Multivariate model adjusted for age, sex, blinatumomab exposure, and bone marrow disease at infusion.
Censored at 24 months because of small sample size beyond 24 months.
![RFS by cytogenetic risk category and individual high-risk lesion. (A) RFS by cytogenetic risk category, defined as the time from onset of remission to relapse in the patients who achieved a complete response. Data were censored for allogeneic HSCT or other alternative therapy during remission (HSCT: n = 7 [high], 3 [intermediate], 4 [favorable], 9 [uninformative]; alternative therapy: n = 5 [high], 1 [intermediate], 7 [favorable], 9 [uninformative]). (B) RFS for KMT2A-rearrangment compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). (C) RFS for Ph+ stratum compared with all others (HSCT: n = 0 [Ph+], 22 [all others]; alternative therapy: n = 4 [Ph+], 19 [all others]). (D) RFS for Ph-like stratum compared with all others (HSCT: n = 6 [Ph-like], 17 [all others]; alternative therapy: n = 1 [Ph-like], 21 [all others]). (E) RFS for infant leukemia with KMT2A rearrangement compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). Tick marks indicate the time of censoring.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/14/10.1182_blood.2021012727/7/m_bloodbld2021012727f1a.png?Expires=1769091192&Signature=ufqMp3zer9sK15ohtef6X~MCLQyMKpC1fSXpGF~xeBKocJd69xfFqRLH-50vAAWmClPIeRzdNP331XibyLbKFCFdzXYRaeB5qQgloTCUHXOhRtjc8lsvXKjU8UaLxvHzu3n4mF8hVQ-nq7hI1qaallvFnhVWTR5rSDbau4D8jkmgok1Ftp8H47FMt4IJYZcvs6GK4tM2GFwcaGLxdp7Sj4ZunqKbe5UQdTEoYTN992MKbZbcITAIJhbY~jNJMifrShyBTkH99tK24BIg6LoEWCqI-9XWN3C8z2CZ-10gtPzaEtimm6CGnmMOIRqMQ6g8eJYFagU3om7j3lKRkREpHw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
![RFS by cytogenetic risk category and individual high-risk lesion. (A) RFS by cytogenetic risk category, defined as the time from onset of remission to relapse in the patients who achieved a complete response. Data were censored for allogeneic HSCT or other alternative therapy during remission (HSCT: n = 7 [high], 3 [intermediate], 4 [favorable], 9 [uninformative]; alternative therapy: n = 5 [high], 1 [intermediate], 7 [favorable], 9 [uninformative]). (B) RFS for KMT2A-rearrangment compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). (C) RFS for Ph+ stratum compared with all others (HSCT: n = 0 [Ph+], 22 [all others]; alternative therapy: n = 4 [Ph+], 19 [all others]). (D) RFS for Ph-like stratum compared with all others (HSCT: n = 6 [Ph-like], 17 [all others]; alternative therapy: n = 1 [Ph-like], 21 [all others]). (E) RFS for infant leukemia with KMT2A rearrangement compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). Tick marks indicate the time of censoring.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/139/14/10.1182_blood.2021012727/7/m_bloodbld2021012727f1b.png?Expires=1769091192&Signature=e7EksCrx85NELNmBnSXnUDhABqEytRR12R8zjTz1cJ1hwataMPeOemTKwK6hYvbo291YzexXzAw~ezrOBqjOqgA4NsLhD~JFRjxoLsgaED3XHu0TTlU2NPjlY0X1~FtZ2Q3LOwMGcjAi75tNB7KOgbTnH-kbQDjctklfoR2TKDUuH3flJlWB4j-MW-voqpv274ZxsPoDJCTea81m105smoeaBmQXk~OovBw8mObiiNOyrm1zDGukG5Pfu2AOg7hZKELRoQte9kWQSPYuT-fMK5VvljXBJXuqC6UQ8YSK6nJJEnPp-Hmb8cCbfwpyt52hQTf0ChVzStA5TC2YidZcsA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
RFS by cytogenetic risk category and individual high-risk lesion. (A) RFS by cytogenetic risk category, defined as the time from onset of remission to relapse in the patients who achieved a complete response. Data were censored for allogeneic HSCT or other alternative therapy during remission (HSCT: n = 7 [high], 3 [intermediate], 4 [favorable], 9 [uninformative]; alternative therapy: n = 5 [high], 1 [intermediate], 7 [favorable], 9 [uninformative]). (B) RFS for KMT2A-rearrangment compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). (C) RFS for Ph+ stratum compared with all others (HSCT: n = 0 [Ph+], 22 [all others]; alternative therapy: n = 4 [Ph+], 19 [all others]). (D) RFS for Ph-like stratum compared with all others (HSCT: n = 6 [Ph-like], 17 [all others]; alternative therapy: n = 1 [Ph-like], 21 [all others]). (E) RFS for infant leukemia with KMT2A rearrangement compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). Tick marks indicate the time of censoring.
RFS by cytogenetic risk category and individual high-risk lesion. (A) RFS by cytogenetic risk category, defined as the time from onset of remission to relapse in the patients who achieved a complete response. Data were censored for allogeneic HSCT or other alternative therapy during remission (HSCT: n = 7 [high], 3 [intermediate], 4 [favorable], 9 [uninformative]; alternative therapy: n = 5 [high], 1 [intermediate], 7 [favorable], 9 [uninformative]). (B) RFS for KMT2A-rearrangment compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). (C) RFS for Ph+ stratum compared with all others (HSCT: n = 0 [Ph+], 22 [all others]; alternative therapy: n = 4 [Ph+], 19 [all others]). (D) RFS for Ph-like stratum compared with all others (HSCT: n = 6 [Ph-like], 17 [all others]; alternative therapy: n = 1 [Ph-like], 21 [all others]). (E) RFS for infant leukemia with KMT2A rearrangement compared with all others (HSCT: n = 0 [KMT2A rearrangement], 22 [all others]; alternative therapy: n = 0 [KMT2A rearrangement], 23 [all others]). Tick marks indicate the time of censoring.
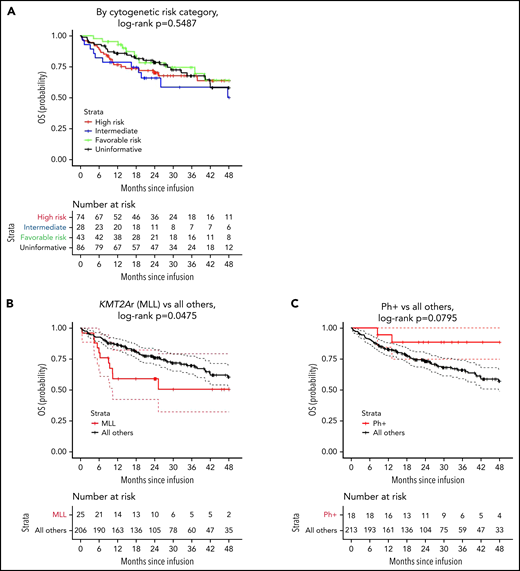
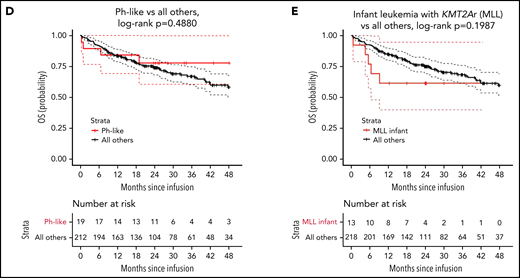
OS by cytogenetic risk category and individual high-risk lesion. (A) OS by cytogenetic risk category, defined as the time from infusion to date of death from any cause. (B) OS for KMT2A rearrangement compared with all others. (C) OS for Ph+ stratum compared with all others. (D) OS for Ph-like stratum compared with all others. (E) OS for infant leukemia with KMT2A rearrangement compared with all others.
OS by cytogenetic risk category and individual high-risk lesion. (A) OS by cytogenetic risk category, defined as the time from infusion to date of death from any cause. (B) OS for KMT2A rearrangement compared with all others. (C) OS for Ph+ stratum compared with all others. (D) OS for Ph-like stratum compared with all others. (E) OS for infant leukemia with KMT2A rearrangement compared with all others.
In a secondary analysis using evidence of MRD as a relapse endpoint, there was no difference in RFS for patients with KMT2A rearrangements compared with all others (P = .3797, supplemental Figure 5A), with a 2-year MRD-RFS of 46% [95% CI, 29-73] for those with KMT2A rearrangements and 56% [95% CI, 49-66] for all others. Similarly, there was no difference in MRD-RFS for those with Ph+ B-ALL compared with all others (P = .2216; 2-year RFS: 71% [95% CI, 53-96] Ph+ and 53% [95% CI, 46-62] all others; supplemental Figure 5B) or for those with Ph-like ALL compared with all others (P = .4759; 2-year RFS: 84% [95% CI, 66-100] Ph-like and 54% [95% CI, 46-62] all others, supplemental Figure 5C). Similar findings were noted in patients with the favorable lesions hyperdiploidy and ETV6/RUNX1 (supplemental Figure 5D-E). In patients with KMT2A rearrangements, there were no MRD relapses. In Ph+ patients, there were 4 MRD relapses, all of whom went on to receive tyrosine kinase inhibitor or cytotoxic treatment within the month, and 3 remain in long-term remissions, and in Ph-like patients, there was 1 MRD relapse who subsequently received alternate therapy and remains in long-term remission.
Thirteen patients with KMT2A-rearranged B-ALL were diagnosed prior to 1 year of age, therefore meeting criteria for infant B-ALL. Two-year RFS for these patients was 67% (95% CI, 45-99, Figure 1E) and OS was 62% (95% CI, 40-95, Figure 2E), with 1 patient who was inevaluable for response due to toxicity-associated death prior to day 28. In a multivariable analysis comparing these patients to all others, adjusting for age, sex, blinatumomab exposure, and bone marrow disease at infusion, there was no increase in risk of relapse (adjusted HR, 0.70; 95% CI, 0.21-2.90; P = .7040; Table 4).
Six patients exhibited myeloid lineage switch following infusion: 5 patients with morphologic relapse and 1 patient who did not respond to initial infusion, and lineage switch was noted on the first postinfusion disease assessment. Five of the patients had KMT2A rearrangements, including the patient with no response, and 3 of those with morphologic relapse had infant ALL. The sixth patient did not have informative cytogenetics. Relapse generally occurred within 6 months, with the exception of the 1 patient with uninformative cytogenetics who remained in remission for 21 months following infusion. All patients who experienced lineage switch died of their disease.
OS
Overall survival was not significantly different between the strata (P = .5488; 2-year OS: 70% [95% CI, 60-82] high risk, 66% [95% CI, 50-87] intermediate, 78% [95% CI, 66-93] favorable, and 79% [95% CI, 70-88] uninformative, Figure 2A). However, the probability of survival for patients with KMT2A rearrangement was statistically lower compared with all others (P = .0475; 2-year OS: 59% [95% CI, 42-82] KMT2A-rearrangment vs 76% [95% CI, 70-83] all others; Figure 2B). There was a trend toward significance for improved overall survival in patients with Ph+ B-ALL (P = .07954; 2-year OS: 88% [95% CI, 75-100] Ph+ vs 73% [95% CI, 67-80] all others; Figure 2C) and no difference between Ph-like and all others (P = .4880; 78% [95% CI, 61-100] Ph-like vs 74% [95% CI, 68-80] all others; Figure 2C). In multivariate analysis, adjusting for age, sex, blinatumomab exposure, and bone marrow disease at infusion, no cytogenetic risk category emerged as associated with OS (Table 3). Multivariable analyses examining individual genomic lesions compared with all others, adjusting for the same variables, found a significant increased risk for KMT2A rearrangement (P = .0212; adjusted HR, 2.59 [95% CI, 1.15-5.81]; Table 4). Neither Ph+ nor Ph-like demonstrated statistically significant associations with OS (Ph+ adjusted HR, 0.41 [95% CI, 0.10-1.71], P = .2211; Ph-like adjusted HR, 0.78 [95% CI, 0.27-2.20], P = .6351; Table 4). In patients with KMT2A-rearranged infant leukemia (n = 13), there was a 3.6-fold increase in risk of all-cause death (95% CI, 1.04-12.75; P = .0434; Table 4).
Discussion
This analysis of children and young adults with relapsed/refractory ALL treated with the CD19-directed CAR T-cell products CTL019, huCART19, and tisagenlecleucel is the first large study to demonstrate similar outcomes across cytogenetic risk categories and extends the findings of smaller studies that suggested similar findings for individual lesions but had few subjects.8,11,39 Patients with high-risk cytogenetic and genomic lesions, including KMT2A-rearrangment, Ph+, Ph-like, hypodiploid, and TCF3/HLF, a group that comprised approximately one-third of the cohort, demonstrated similar rates of CR, RFS, and OS to patients categorized as having intermediate, favorable, or uninformative genetic characteristics. Further, multivariable analysis of RFS, controlling for age, sex, blinatumomab exposure,10,40-42 and bone marrow disease at infusion, did not demonstrate a significant increase in risk of relapse or death for the high-risk cytogenetics group. Perhaps revealingly, there was similarly no difference in baseline disease characteristics across cytogenetic risk groups, with a majority in second or greater relapse (49% high risk, 55% others), 41% with refractory disease (39% others), and half with a history of HSCT (51% high risk, 39% others).
Total relapse rate for the entire cohort was 34%, with no difference between cytogenetic groups, and CD19+ and CD19− relapse rates occurred with similar frequencies in the different groups, although myeloid lineage switch occurred primarily in patients with KMT2A rearrangements. Six cases of myeloid lineage switch following CAR T-cell infusion were seen, 5 in KMT2A-rearranged cases and 1 with uninformative cytogenetics, all of whom subsequently succumbed to their disease. This finding is consistent with prior observations that lineage switch is a rare phenomenon associated with poor prognosis43 and thought to occur most commonly with specific genetic subtypes that have greater plasticity, such as KMT2A-rearrangement, although not exclusively.12,13 CD19-directed CAR T-cell immune pressure inducing lineage switch as a mechanism of CAR resistance has been demonstrated in murine models44 as well as in prior clinical trials.8 Our data, with a 21% incidence of lineage switch among KMT2A-rearranged leukemia relapses, agrees with that of prior work8 and suggests that relapse occurs rapidly in the context of KMT2A-mediated lineage switch, conferring a dismal prognosis. This finding, however, is balanced against the encouraging outcomes demonstrated overall in KMT2A-rearranged B-ALL and those seen in KMT2A-associated infant B-ALL, and future work should focus on determining if there are baseline or modifiable features that can predict or avoid lineage switch in this population.
KMT2A translocations are known to be associated with chemo-refractory disease that has a higher likelihood of relapse, resulting in poor outcomes in children and young adults and conferring a dismal prognosis for infants.45 Despite advancements in overall cure rates for childhood leukemia, there has been only modest improvement for patients with KMT2A-rearranged ALL with EFS and OS clustering around 50%, relapses rates between 50% to 60%, and salvage following relapse very challenging.46-48 The RFS observed with CAR T-cell therapy in this study, a 2-year RFS of 46%, is encouraging for patients with relapsed/refractory B-ALL with KMT2A rearrangements, as is the lack of statistically significant difference in RFS compared with other B-ALL subtypes; however, it may be difficult to detect a difference in this relatively small subset. There was a non–statistically significant trend toward increased risk for relapse in multivariable analysis, a finding that warrants confirmation with a larger sample size and suggests that factors for which multivariable analyses adjust may contribute to these outcomes. We note that a large proportion (56%) of patients with KMT2A-rearranged ALL were in an MRD− remission at the time of infusion, either because relapse presented in an isolated extramedullary site or due to bridging chemotherapy, suggesting chemo-sensitive disease. As has been found in other KMT2A-driven disease subtypes, relapses tended to occur within the first year.49 Additionally, patients who relapse after CAR T-cell therapy, unfortunately, largely go on to die of their disease, with a 2-year OS of 59% compared with 76% for all others, with multivariable analysis showing a 2.6-fold increase in risk of all-cause death. Nevertheless, these outcomes for KMT2A-rearranged B-ALL treated with CAR T cells are promising, particularly in light of the fact that 68% of these patients had 2 or more prior relapses.
The outcome for infants with KMT2A-rearranged ALL treated with current frontline intensive regimens are even worse than those of older children, with 5-year EFS rates of about 35% and OS rates of about 45% reported in 4 major studies,48,50-52 without a clear benefit to HSCT. Relapse unfortunately portends a dismal prognosis, with a 3-year OS rate of 24% on Interfant-99.53 RFS after CAR T-cell therapy was 67% at 2 years for 13 patients with infant B-ALL with KMT2A rearrangement included in the current study, 8 of them in second or greater relapse (62%), and 8 who had relapsed after HSCT (62%). Although limited by small sample size, these data are encouraging and suggest that patients with KMT2A-rearranged infant B-ALL treated with CD19 CAR T cells are not at higher risk of relapse than others. However, they are at a more than threefold increased risk of all-cause death, perhaps related to the challenge of salvaging these relapses.
In an unadjusted analysis, Ph+ ALL appears to be associated with improved RFS, although this effect is dampened when using MRD-level disease to define relapse, possibly related to the ability to detect relapse earlier with BCR-ABL1 polymerase chain reaction. Moreover, in multivariate analysis, there was no statistically significant difference in relapse risk when adjusted for disease burden, potentially related to the higher proportion (56%) of patients with <0.01% MRD preinfusion. Interestingly, neither the KMT2A-rearranged nor Ph-like RFS analyses changed with the use of MRD-level disease as a relapse endpoint, despite the fact that disease monitoring intervals are the same. Differences in disease kinetics or the sensitivity and specificity of disease monitoring by lesion-specific polymerase chain reaction may account for this effect.
It should be noted that this study used a clinically pragmatic approach to collecting and categorizing cytogenetic data, relying on disease assessments that had been performed at diagnosis and relapse, and retrospectively abstracting genetic information from the resultant reports. This presents 2 challenges: First, that not all patients may have had the full complement of leukemia genetic testing, although KMT2A rearrangements, ETV6/RUNX1 fusion, BCR/ABL1 fusion, and hypo/hyperdiploidy are routinely screened for, and further, that the determination of specific lesions and their other partners are dependent on the sensitivity of specific tests. Second, the available cytogenetic analysis does not necessarily reflect the disease at the time of CAR T-cell infusion, although data suggests that sentinel lesions are largely preserved in relapse,6,34-36 and the acquisition of many of the high-risk sentinel lesions is rare.54,55 The selection of cytogenetic lesions for this study was based on the genetic aberrations that are most routinely screened for in clinical practice, with the aim of limiting missing data and maximizing clinical applicability of the results. However, the authors acknowledge that the interplay of genetic features is complex, and there is a need to understand these features with more granularity. Further investigation is required to examine the role of newly described sentinel cytogenetic lesions and the more widespread use of sequencing to identify genomic mutations such as TP53 alterations, NR3C1/BTG1 deletions, and RAS mutations,6 as well as those nondriver lesions thought to act in concert with others.28
In conclusion, this study demonstrates that CTL019/huCART19 therapy produces durable remissions of relapsed/refractory ALL with similar CR, RFS, and OS rates across cytogenetic risk groups. Prediction models for relapse after CAR T-cell therapy and therapies to reduce the risk of relapse remain important avenues for future research to improve outcomes.
Acknowledgments
The authors thank the Cancer Immunotherapy Program clinical research team at the Children’s Hospital of Philadelphia.
This study was supported by the Children’s Hospital of Philadelphia Frontier Program. The clinical trials included in this post hoc pooled analysis were supported by clinical trial awards funded by Novartis Pharmaceuticals and a research alliance between the University of Pennsylvania, Novartis Pharmaceuticals, and the Children’s Hospital of Philadelphia Frontier Program. A.B.L., S.A.G., and S.L.M. are supported by grants from the National Institutes of Health (5K12CA076931–22 to A.B.L. and 5P01CA214278–04 to S.A.G. and S.L.M.).
Authorship
Contribution: A.B.L., K.J.D., S.P.H., and S.L.M. were involved in the conception, design, and planning of the study. A.B.L., K.J.D., R.M., A.D., L.W., S.R.R., C.C., D.M.B., M.P., H.N., S.A.G., D.M.B., and S.L.M. collected the data. Y.L. and H.L. did the statistical analysis. All authors reviewed the data analyses, contributed to data interpretation and writing of the report, and approved the final version of the submitted report.
Conflict-of-interest disclosure: C.C has served as a consultant for Novartis Pharmaceuticals. S.P.H. has received consulting fees from Novartis, honoraria from Jazz Pharmaceuticals and Amgen, and owns common stock in Amgen. S.A.G. has received research and/or clinical trial support from Novartis, Servier, and Kite and has participated in consulting, study steering committees, or scientific/clinical advisory boards for Novartis, Cellectis, Adaptimmune, Eureka, TCR2, Juno, GlaxoSmithKline, Vertex, Cure Genetics, Humanigen, and Roche. S.R.R. has research funding and has served as a consultant form Pfizer, Inc. S.L.M has served as a consultant for Novartis Pharmaceuticals, Kite Pharma, and Wugen and receives clinical trial funding from Novartis Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Allison Barz Leahy, Children’s Hospital of Philadelphia, Colket Translational Building, 3501 Civic Center Blvd, Philadelphia, PA 19104; e-mail: barza@chop.edu; Shannon Maude, Children’s Hospital of Philadelphia, Colket Translational Building, 3501 Civic Center Blvd, Philadelphia, PA 19104; e-mail: maude@chop.edu.
Individual participant data that underlie the results reported in this article, after deidentification, may be shared with investigators. Data will only be shared with investigators who provide a methodologically sound proposal with approved aims, as long as release of that data does not compromise an ongoing trial or study, that there is a strong scientific rationale for the data to be used for the requested purpose, and that investigators who have invested time and effort into developing these trials have a period of exclusivity in which to pursue their own aims with the data, before key trial data are available to others. Proposals should be directed to the corresponding author, and to gain access, data requestors will need to sign a data access agreement.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal