

Key Points
Primary PCL displays a specific genomic and transcriptomic profile when compared with newly diagnosed myeloma.
Primary PCL with t(11;14) is a distinct genomic and transcriptional entity, with better overall survival.

Abstract
Primary plasma cell leukemia (pPCL) is an aggressive form of multiple myeloma (MM) that has not benefited from recent therapeutic advances in the field. Because it is very rare and heterogeneous, it remains poorly understood at the molecular level. To address this issue, we performed DNA and RNA sequencing of sorted plasma cells from a large cohort of 90 newly diagnosed pPCL and compared with MM. We observed that pPCL presents a specific genomic landscape with a high prevalence of t(11;14) (about half) and high-risk genomic features such as del(17p), gain 1q, and del(1p32). In addition, pPCL displays a specific transcriptome when compared with MM. We then wanted to characterize specifically pPCL with t(11;14). We observed that this subentity displayed significantly fewer adverse cytogenetic abnormalities. This translated into better overall survival when compared with pPCL without t(11;14) (39.2 months vs 17.9 months, P = .002). Finally, pPCL with t(11;14) displayed a specific transcriptome, including differential expression of BCL2 family members. This study is the largest series of patients with pPCL reported so far.
Introduction
Plasma cell leukemia (PCL) is a rare and aggressive form of multiple myeloma (MM), defined by the presence of >20% of plasma cells in the peripheral blood and/or a plasma cell count greater than 2.109/L.1 It represents 2% to 4% of multiple myeloma cases (ie, 0.04 cases per 100 000 people per year).2 It is said to be primary (pPCL) if it occurs de novo or secondary (sPCL) if it corresponds to a leukemic transformation of a previously known myeloma. pPCL accounts for 60% to 70% of PCL.
Compared with MM, pPCL displays distinct biological and clinical features,3,4 such as a younger age at diagnosis, a more aggressive presentation with frequent extramedullary diseases, and fewer lytic bone lesions. Despite the improvement of MM therapy, pPCL continues to have a poor prognosis, with a median overall survival between 18 and 36 months.5-8
Given the low incidence and the heterogeneity of pPCL, biological knowledge is lacking, especially the molecular process responsible for its aggressiveness. To address this issue, we took advantage of a large series of pPCL to describe the genomic and transcriptomic landscape of pPCL. Given the high incidence of t(11;14) that we and others9,10 observed in this disease, we aimed to determine if this subgroup displayed a specific genomic profile and a better clinical outcome.
Study design
Between 2014 and 2020, bone marrow samples were collected from 90 newly diagnosed pPCL patients within hospitals involved in the Intergroupe Francophone du Myélome and shipped overnight to Toulouse central laboratory. All patients provided informed consent. This study was approved by the Toulouse Ethics Committee. Plasma cells were sorted from bone marrow samples using CD138 immunomagnetic cell sorting (>90% purity). DNA and RNA sequencing were performed using NextSeq 500 (Illumina). Data obtained from pPCL samples were compared with those from to 907 and 288 newly diagnosed MM, respectively. Technical details of sequencing and bioinformatics analysis are available in supplemental Method.
To evaluate overall survival of pPCL patients according to t(11;14) status, we selected 128 patients for whom clinical data were available, diagnosed between 2000 and 2020, followed up for >18 months or having died of pPCL, and for whom t(11;14) was established by next-generation sequencing (53 patients out of 90 from sequencing cohort) or by fluorescence in situ hybridization as previously described (75 additional patients) (supplemental Figure 1). Clinical data and treatment regimens of these 128 patients are presented in supplemental Table 1. Survival rate was estimated by the Kaplan-Meier method. Tests were 2-sided, and P values <.05 were considered significant. Statistical analysis was conducted using R.
Results and discussion
Genome analysis on 90 patients highlighted a specific genomic profile of pPCL (Figure 1A). Among the cytogenetic abnormalities, pPCL patients displayed higher incidences of t(11;14) (51.1% vs 23.1%, P < .001) and t(14;16) (14.4% vs 3.4%, P < .001) but an identical incidence of t(4;14) (11.1% vs 9.8%, P = .7). Hyperdiploidy was less frequent in pPCL compared with MM (20.0% vs 56.9%, P < .0001), with less trisomy 5 (7.8% vs 44.4%, P < .0001). We observed that pPCL presented higher incidence of adverse secondary lesions compared with MM, such as deletion 17p (30.0% vs 9.5%, P < .0001; including 12.2% vs 3.1% of TP53 bi allelic inactivation, P < .0001) and gain 1q (53.3% vs 31.9%, P < .001; including 11.1% vs 5.3% of amplification 1q, P < .05) but also deletion 1p32 (24.4% vs 9.4%, P < .001). Our results are consistent with previous reports on smaller series.9,11-16 Despite their heterogeneity, pPCL present a specific genomic landscape with a high prevalence of t(11;14) (about half) and high-risk genomic features.
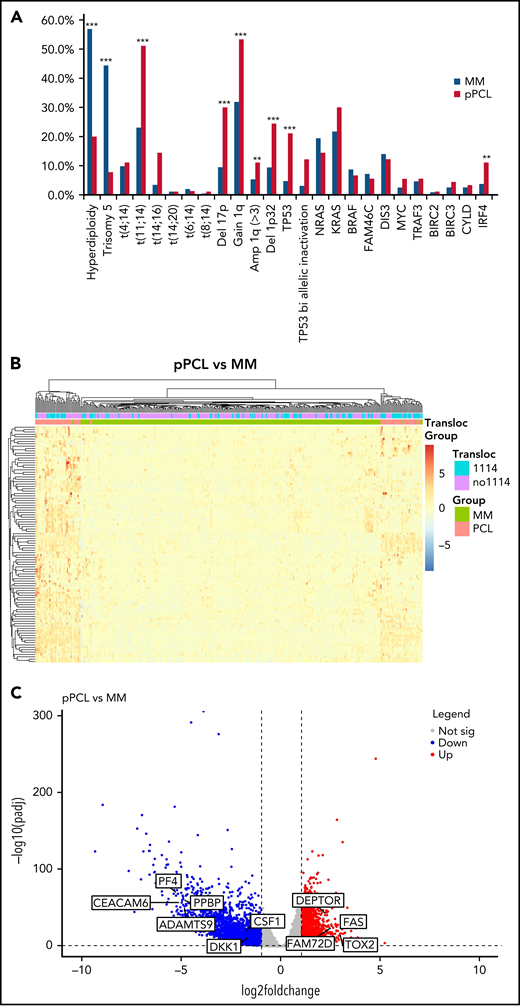
Genomic and transcriptome of malignant plasma cells from primary PCL compared with newly diagnosed multiple myeloma. (A) Incidence of main cytogenetics abnormalities and mutations in 90 primary PCL compared with 907 newly diagnosed MM, ***P < .0001, **P < .005. (B) Unsupervised clustering of 90 primary PCL and 288 newly diagnosed MM based on the expression levels of the 100 most variable genes. (C) Gene expression profiling comparing differentially expressed genes between primary PCL and newly diagnosed MM, with at least twofold change (P < .05, FDR<0.01). (D) GSEA from 90 primary PCL compared with 288 newly diagnosed MM.
Genomic and transcriptome of malignant plasma cells from primary PCL compared with newly diagnosed multiple myeloma. (A) Incidence of main cytogenetics abnormalities and mutations in 90 primary PCL compared with 907 newly diagnosed MM, ***P < .0001, **P < .005. (B) Unsupervised clustering of 90 primary PCL and 288 newly diagnosed MM based on the expression levels of the 100 most variable genes. (C) Gene expression profiling comparing differentially expressed genes between primary PCL and newly diagnosed MM, with at least twofold change (P < .05, FDR<0.01). (D) GSEA from 90 primary PCL compared with 288 newly diagnosed MM.
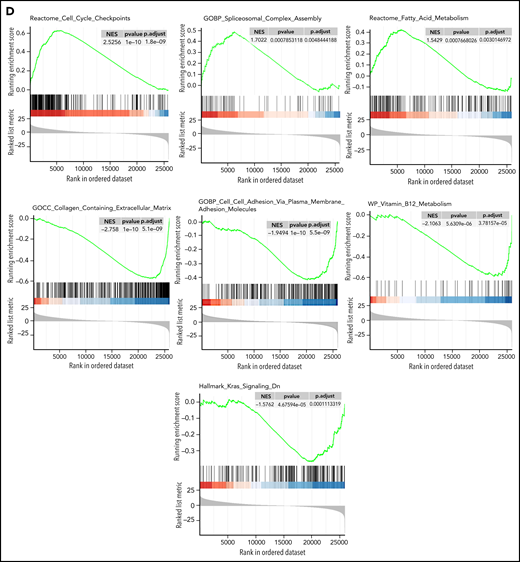
RNA sequencing analysis also highlighted the specificity of pPCL. Indeed, unsupervised hierarchical clustering analysis showed 2 distinctive cluster groups between pPCL (n = 80) and MM (n = 288) (Figure 1B). We found 2835 genes upregulated and 4070 downregulated in pPCL. Among top upregulated genes, we observed FAM72D, recently identified as a 1q21 marker of proliferation,17 and TOX2, which belongs to the 15-gene prognostic classifier described by the Intergroupe Francophone du Myélome18 (Figure 1C). Interestingly, we also observed a downregulation of the inhibitor of osteoblastic differentiation DKK1 and the osteoclast activating factor M-CSF. Gene set enrichment analysis (GSEA) revealed an upregulation of genes involved in the cell cycle, fatty acid metabolism, and spliceosome and a downregulation of genes involved in adhesion, extracellular matrix, KRAS signaling, and B12 vitamin metabolism (Figure 1D). These biological differences may explain the distinct clinical features such as more proliferating presentation and more diffusion to extramedullary sites in pPCL but fewer bone lytic lesions. Despite the small size of previous pPCL cohorts, upregulation of genes involved in the spliceosome was also recently described,10 and downregulation of adhesion molecules and extracellular matrix proteins too.15
Given the large frequency of t(11;14) in pPCL, we aimed at specifically characterizing this subgroup. Interestingly, pPCL with t(11;14) tended to display more TP53 mutations (34.0% vs 16.0%, P = .054), but this was associated to a deletion 17p only in half cases, whereas all TP53 mutations for the non-t(11;14) subgroup were biallelic inactivation. Importantly, pPCL with t(11;14) had fewer adverse cytogenetic abnormalities such as gain 1q (29.7% vs 79.1%, P < .0005), including amplification 1q (2.1% vs 20.9%, P < .01), and deletion 1p32 (8.5% vs 41.9%, P < .001) (Figure 2A). Only 10% of the pPCL with t(11;14) had at least 2 lesions among deletion 17p, gain 1q, and deletion 1p32, whereas it was 46% for the non-t(11;14) subgroup (P < .0005). In addition, t(4;14) and t(14;16) pPCL were by definition exclusively present in the non-t(11;14) subgroup. As expected, all these differences translated into a better outcome because median overall survival for pPCL patients with t(11;14) was significantly longer than those without t(11;14) (39.2 months vs 17.9 months, respectively, P = .002) (Figure 2B). Gene expression profile showed that pPCL with and without t(11;14) clustered in 2 distinct groups (Figure 2C). We found 875 genes upregulated and 1214 downregulated in pPCL with t(11;14). As expected, among the most upregulated genes was CCND1. As recently shown by Todoerti et al,19 several members of the BCL2 family genes were also significantly differentially expressed between pPCL subgroups. We observed that PMAIP1 and BCL2L15 were overexpressed and BCL2L1 and BAK1 underexpressed. Although a fold change <2, BCL-2 and BCL2-L2 were also overexpressed and BAX underexpressed when compared with non-t(11;14) subgroup. Furthermore, as recently shown in MM with t(11;14),19,20 pPCL with t(11;14) displayed an upregulation of immature B-cell markers PAX5 and CD79A (Figure 2D), supporting the BCL2 dependency and suggesting a distinctive oncogenic mechanism for this subgroup. GSEA also revealed a downregulation of genes involved in the cell cycle, NRAS and KRAS pathways, adhesion, and extracellular matrix (Figure 2E). Of note, although a small cluster of pPCL with t(11;14) was present in the same branch as the non-t(11;14) subgroup, BCL2 members family and CCND1 expression and outcome were similar as the larger cluster of pPCL with t(11;14). Globally, we highlighted that pPCL with t(11;14) is a distinguishable entity, which may notably be less proliferative, in accordance to a better prognosis, but probably more BCL2 dependent. Venetoclax, a BCL2 inhibitor, had evidence of efficacy in t(11;14) MM.21,22 Our results are consistent with the efficacy of Venetoclax also described in PCL patients23,24 and support its evaluation in clinical trials, especially for patients with t(11;14).
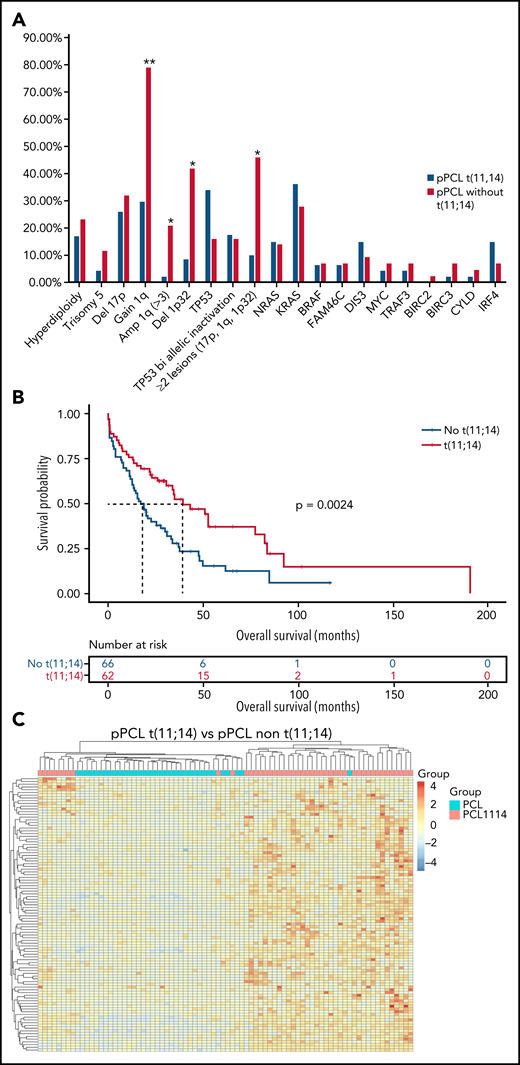
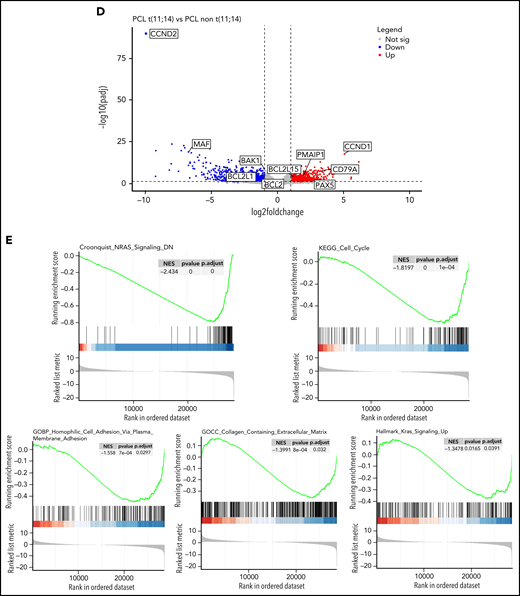
Genomic and transcriptome of malignant plasma cells from primary PCL displaying t(11;14) compared with primary PCL without t(11;14). (A) Incidence of main cytogenetics abnormalities and mutations in 46 primary PCL displaying t(11;14) compared with 44 primary PCL without t(11;14), **P < .0001, *P < .01. (B) Kaplan-Meyer overall survival of primary PCL patients according to t(11;14). (C) Unsupervised clustering of 46 primary PCL displaying t(11;14) and 44 primary PCL without t(11;14) based on the expression levels of the 100 most variable genes. (D) Gene expression profiling comparing differentially expressed genes between primary PCL displaying t(11;14) and primary PCL without t(11;14), with at least twofold change (P < .05, FDR < .01). (E) GSEA from 46 primary PCL displaying t(11;14) and 44 primary PCL without t(11;14). FDR, false discovery rate.
Genomic and transcriptome of malignant plasma cells from primary PCL displaying t(11;14) compared with primary PCL without t(11;14). (A) Incidence of main cytogenetics abnormalities and mutations in 46 primary PCL displaying t(11;14) compared with 44 primary PCL without t(11;14), **P < .0001, *P < .01. (B) Kaplan-Meyer overall survival of primary PCL patients according to t(11;14). (C) Unsupervised clustering of 46 primary PCL displaying t(11;14) and 44 primary PCL without t(11;14) based on the expression levels of the 100 most variable genes. (D) Gene expression profiling comparing differentially expressed genes between primary PCL displaying t(11;14) and primary PCL without t(11;14), with at least twofold change (P < .05, FDR < .01). (E) GSEA from 46 primary PCL displaying t(11;14) and 44 primary PCL without t(11;14). FDR, false discovery rate.
In summary, we performed a study on the largest series of patients with pPCL reported so far. Using high-throughput technologies, we demonstrated that pPCL had a specific genomic and transcriptomic landscape that may explain its biological and clinical features. It would be useful to extend this analysis to pPCL defined from 5% of circulating plasma cells.25 We also described the biological and clinical specificities of t(11;14) pPCL, supporting the use of novel therapeutic approaches such as BCL2 inhibitors, but prospective studies are necessary to confirm it.
Acknowledgments
The authors thank the Intergroupe Francophone du Myélome for providing patient samples and clinical data.
This work was supported by the National Institutes of Health, National Cancer Institute grants PO1-155258 and P50-100707 (H.A.L.) and the Cancer Pharmacology of Toulouse and Region (CAPTOR) program. The Centre de Recherches en Cancérologie de Toulouse Team 13 is supported by the Fondation ARC (Association pour la Recherche sur la Cancer, grant PGA1*20160203788).
Authorship
Contribution: J.C. and H.A.-L. designed the research and analyzed data; T.C., L.B., S.M., and A.S., performed the analysis; L.D.S.F., R.L., and L.P. performed the bioinformatics analysis; T.C., J.C., and H.A.-L. wrote the manuscript, which was reviewed and edited by the other coauthors; and all other authors provided study samples and clinical data.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jill Corre, Myeloma Oncogenesis Laboratory, IUC-Oncopole, 1 avenue Irene Joliot-Curie, 31059 Toulouse, France; e-mail: corre.jill@iuct-oncopole.fr; and Hervé Avet-Loiseau, Myeloma Oncogenesis Laboratory, IUC-Oncopole, 1 avenue Irene Joliot-Curie, 31059 Toulouse, France; e-mail: Avetloiseau.herve@iuct-oncopole.fr.
Data for this manuscript can be found in the Gene Expression Omnibus database under accession number PRJNA804686.
Requests for data sharing may be submitted to Jill Corre and Hervé Avet-Loiseau (corre.jill@iuct-oncopole.fr and Avetloiseau.herve@iuct-oncopole.fr).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal