

Key Points
Transplant outcomes in older patients with AML in first remission are primarily determined by characteristics present at diagnosis.
MRD in older patients with AML is associated with high-risk baseline features and does not affect LFS independent of baseline risk.

Abstract
Older patients with acute myeloid leukemia (AML) have high relapse risk and poor survival after allogeneic hematopoietic cell transplantation (HCT). Younger patients may receive myeloablative conditioning to mitigate relapse risk associated with high-risk genetics or measurable residual disease (MRD), but older adults typically receive reduced-intensity conditioning (RIC) to limit toxicity. To identify factors that drive HCT outcomes in older patients, we performed targeted mutational analysis (variant allele fraction ≥2%) on diagnostic samples from 295 patients with AML aged ≥60 years who underwent HCT in first complete remission, 91% of whom received RIC, and targeted duplex sequencing at remission in a subset comprising 192 patients. In a multivariable model for leukemia-free survival (LFS) including baseline genetic and clinical variables, we defined patients with low (3-year LFS, 85%), intermediate (55%), high (35%), and very high (7%) risk. Before HCT, 79.7% of patients had persistent baseline mutations, including 18.3% with only DNMT3A or TET2 (DT) mutations and 61.4% with other mutations (MRD positive). In univariable analysis, MRD positivity was associated with increased relapse and inferior LFS, compared with DT and MRD-negative mutations. However, in a multivariable model accounting for baseline risk, MRD positivity had no independent impact on LFS, most likely because of its significant association with diagnostic genetic characteristics, including MDS-associated gene mutations, TP53 mutations, and high-risk karyotype. In summary, molecular associations with MRD positivity and transplant outcomes in older patients with AML are driven primarily by baseline genetics, not by mutations present in remission. In this group of patients, where high-intensity conditioning carries substantial risk of toxicity, alternative approaches to mitigating MRD-associated relapse risk are needed.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 3559.
Disclosure
Laurie Barclay, MD, has disclosed the following relevant financial relationships: stock, stock options, or bonds: AbbVie Inc. (former).
Learning objectives
Upon completion of this activity, participants will:
- 1.
Describe clinical and genetic determinants of posttransplant leukemia-free survival in older patients with acute myeloid leukemia (AML), according to a targeted mutational genomic analysis
- 2.
Determine molecular genetics of complete remission and minimal residual disease associations with baseline characteristics and posttransplant outcomes in older patients with AML, according to a targeted mutational genomic analysis of paired diagnostic and available remission specimens
- 3.
Identify clinical implications of factors that drive outcomes of allogeneic hematopoietic cell transplantation for AML in older patients, according to a targeted mutational genomic analysis of paired diagnostic and available remission specimens
Release date: June 16, 2022; Expiration date: June 16, 2023
Introduction
Acute myeloid leukemia (AML) in adults aged ≥60 years is associated with inferior outcomes in comparison with younger patients.1-3 In older patients, AML frequently evolves from antecedent myelodysplastic syndromes (MDS),4,5 is enriched for high-risk cytogenetic abnormalities,6 and is often resistant to conventional chemotherapy.4,6,7 Allogeneic hematopoietic cell transplantation (HCT) is the only curative treatment option for many older patients with AML, but prognosis after transplant in this age group is limited.8,9
With current treatment approaches, the inferior outcomes for older patients who undergo transplants for AML are primarily related to a high rate of relapse after transplantation.8 This is related in part to the use of less intensive conditioning regimens for older adults at increased risk of treatment-related toxicity,10,11 but may also reflect enrichment of high-risk biological features in older AML populations.4,12 Genomic assessment of AML cohorts, either at diagnosis or first complete remission (CR1), may identify prognostic subgroups with distinct risks of relapse or nonrelapse mortality (NRM), thereby suggesting specific risk-adapted therapeutic strategies.
Molecular assessment at remission could be particularly meaningful in the older AML population, given recent findings that measurable residual disease (MRD), defined as detectable AML mutations at the time of complete remission (CR), is associated with increased relapse risk among those who undergo reduced intensity conditioning (RIC) regimens.13 These results, however, were from a randomized trial that excluded patients >65 years of age and required that all patients be eligible for myeloablative conditioning (MAC) and thus may not apply equally to a broad population of real-world older patients with AML. At many centers, transplants are offered to patients into their 70s, and reduced-intensity regimens are employed in more than 85% of cases.14
Dedicated studies in older patients with AML may identify those most likely to benefit from transplantation and enable development of strategies tailored to a patient’s specific profile of relapse and toxicity risk, as determined by pretransplant characteristics. In the absence of randomized data in the older AML population, retrospective analysis of real-world older cohorts may be preferable to extrapolation from studies of younger patients. We report a genomic analysis of paired diagnostic and available remission specimens in a multi-institution cohort of older patients with AML who underwent allogeneic transplantation in morphologic CR1.
Methods
Patients and samples
Patients who were diagnosed with AML at age ≥60 and underwent allogeneic transplantation in CR1, with or without hematologic recovery (CR or CR with incomplete recovery [CRi]) at 1 of 6 participating centers (Dana-Farber Cancer Institute, Johns Hopkins University, Memorial Sloan Kettering Cancer Center, The Ohio State University, Roswell Park Comprehensive Cancer Center, and the University of Pennsylvania) were eligible for inclusion (supplemental Table 1 available on the Blood Web site). Patients with acute promyelocytic leukemia or isolated extramedullary disease were excluded. Conditioning regimens and graft-versus-host disease prophylaxis strategies were administered according to the discretion of the treating physician. A total of 295 patients met the inclusion criteria and had a banked bone marrow or peripheral blood sample collected before the initiation of induction chemotherapy. Of those, 192 (65.1%) also had a sample collected in CR1 before transplant. The median age at the time of AML diagnosis was 66 years (range, 60-76). The median follow-up time for survivors was 44 months (range, 6.7-155.3), median time from diagnosis to transplant was 4.8 months (range, 1.9-21), and median time to relapse after transplant was 4.8 months (range, 1-60.8). For patients with remission samples, the median time from remission sample collection to transplant was 2.5 months (range, 0.1-14). Most patients in the remission cohort (121 of 192) received consolidation after CR1 and before transplantation. Five of the 40 patients in the remission cohort with a FLT3-internal tandem duplication (ITD) received a FLT3 inhibitor as part of their consolidation therapy. Additional characteristics of the 295 included patients are reported in Table 1, and a comparison of baseline characteristics between those with and without a remission sample is shown in supplemental Table 2. The study was conducted with the approval of and with waivers of consent from the institutional review boards at the respective institutions.
Cohort characteristics
| Patients | ||
|---|---|---|
| Characteristic | n | % |
| Full cohort, n | 295 | 100 |
| Recipient age, median (range) | 66 (6-76) | |
| Recipient sex | ||
| Female | 117 | 40 |
| Male | 178 | 60 |
| HCT-CI score | ||
| 0 | 83 | 28 |
| 1-2 | 81 | 27 |
| 3+ | 120 | 40 |
| Missing | 11 | 4 |
| Type of AML (clinically defined) | ||
| De novo | 173 | 59 |
| Secondary | 91 | 31 |
| Therapy-related | 31 | 11 |
| Cytogenetics* | ||
| Normal | 136 | 46 |
| Core binding factor | 6 | 2 |
| Complex karyotype | 41 | 14 |
| Other | 112 | 38 |
| 2017 ELN risk group | ||
| Favorable | 53 | 18 |
| Intermediate | 85 | 29 |
| Adverse | 152 | 52 |
| Missing | 5 | 2 |
| Initial therapy | ||
| Intensive induction | 249 | 84 |
| Non-intensive induction | 46 | 16 |
| Reinduction | ||
| Yes | 90 | 31 |
| No | 204 | 69 |
| Missing | 1 | 0.3 |
| Remission quality | ||
| CR with hematologic recovery | 225 | 75 |
| CRi | 67 | 23 |
| Missing | 1 | 0.3 |
| Donor type | ||
| Matched related | 54 | 18 |
| Matched unrelated | 154 | 52 |
| Mismatch related | 7 | 2 |
| Mismatch unrelated | 29 | 10 |
| Haploidentical | 51 | 17 |
| Conditioning regimen | ||
| Myeloablative | 28 | 9 |
| Reduced intensity | 267 | 91 |
| T-cell depletion | 25 | 9 |
| Stem cell source | ||
| Peripheral blood | 216 | 73 |
| Bone marrow | 71 | 24 |
| Umbilical cord blood | 8 | 3 |
| Patients | ||
|---|---|---|
| Characteristic | n | % |
| Full cohort, n | 295 | 100 |
| Recipient age, median (range) | 66 (6-76) | |
| Recipient sex | ||
| Female | 117 | 40 |
| Male | 178 | 60 |
| HCT-CI score | ||
| 0 | 83 | 28 |
| 1-2 | 81 | 27 |
| 3+ | 120 | 40 |
| Missing | 11 | 4 |
| Type of AML (clinically defined) | ||
| De novo | 173 | 59 |
| Secondary | 91 | 31 |
| Therapy-related | 31 | 11 |
| Cytogenetics* | ||
| Normal | 136 | 46 |
| Core binding factor | 6 | 2 |
| Complex karyotype | 41 | 14 |
| Other | 112 | 38 |
| 2017 ELN risk group | ||
| Favorable | 53 | 18 |
| Intermediate | 85 | 29 |
| Adverse | 152 | 52 |
| Missing | 5 | 2 |
| Initial therapy | ||
| Intensive induction | 249 | 84 |
| Non-intensive induction | 46 | 16 |
| Reinduction | ||
| Yes | 90 | 31 |
| No | 204 | 69 |
| Missing | 1 | 0.3 |
| Remission quality | ||
| CR with hematologic recovery | 225 | 75 |
| CRi | 67 | 23 |
| Missing | 1 | 0.3 |
| Donor type | ||
| Matched related | 54 | 18 |
| Matched unrelated | 154 | 52 |
| Mismatch related | 7 | 2 |
| Mismatch unrelated | 29 | 10 |
| Haploidentical | 51 | 17 |
| Conditioning regimen | ||
| Myeloablative | 28 | 9 |
| Reduced intensity | 267 | 91 |
| T-cell depletion | 25 | 9 |
| Stem cell source | ||
| Peripheral blood | 216 | 73 |
| Bone marrow | 71 | 24 |
| Umbilical cord blood | 8 | 3 |
Shown are the pretransplant characteristics of the 295 patients included in the cohort. HCT-CI: hematopoietic cell transplant comorbidity index score. CRi denotes complete remission with incomplete recovery of at least 1 hematopoietic cell lineage.
ELN, European Leukemia Network.
Core binding factor: inv(16) or t(8;21); complex karyotype: 3 or more chromosomal abnormalities within a single clone.
Genetic studies
For diagnostic samples, we performed targeted sequencing of 113 genes known to be recurrently mutated in AML or in germline syndromes predisposing to development of myeloid malignancies (supplemental Table 3). For remission samples, we performed duplex unique molecular identifier-tagged targeted sequencing of 76 genes recurrently mutated in myeloid malignancies (supplemental Table 4). We classified variants as pathogenic on the basis of accepted genetic criteria.15 Annotation of mutations was blinded to clinical characteristics and genetic analysis was locked before merging with clinical data. For remission samples, we performed initial mutation annotation blinded to diagnostic sequencing results, then assessed each remission sample for the presence or absence of mutations present in the paired diagnostic sample (supplemental Table 5).
Statistical analysis
The primary end point was leukemia-free survival (LFS), defined as the time from transplantation until death or relapse of the original AML, whichever occurred first. Overall survival (OS) was defined as the time from transplantation until death from any cause or until censoring at the time last known to be alive. OS and LFS were estimated by the Kaplan-Meier method, and the difference was determined with log-rank tests. Cumulative incidences of NRM and relapse were estimated in competing risk frameworks that treated relapse and NRM as competing events and were compared using the Gray test. Multivariable analysis was performed with Cox models for OS and LFS and Fine and Gray models for NRM and relapse. Risk groupings were derived from the results of univariable and multivariable models. Additional details are provided in the supplemental methods.
Results
Pretreatment genetic characteristics
Sequencing of diagnostic (pre-induction) samples showed that high-risk genetic characteristics were common in this older cohort. Somatic mutations that indicate evolution from antecedent MDS, which are associated with poor outcome,4 were present in 127 of the 295 patients (43.1%); TP53 mutations and FLT3-internal tandem duplications (FLT3-ITDs) without concurrent NPM1 mutations, both of which reflect adverse molecular risk, were each present in 33 (11.2%); and NPM1 mutations without concurrent FLT3-ITDs, which are associated with favorable transplantation outcomes in younger patients, were present in 36 (12.2%; Figure 1).4,12,16FLT3-ITDs with concurrent NPM1 mutations were present in 29 patients (9.8%). Tyrosine kinase inhibitors were used after transplantation in 22 patients with FLT3-ITDs.
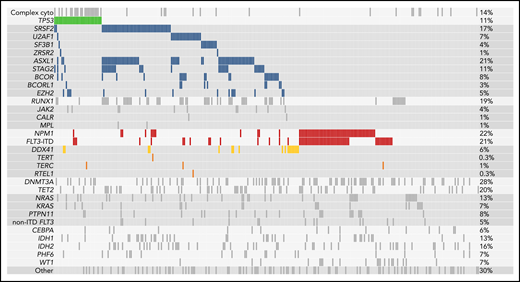
Mutations present at the time of diagnosis. Shown are all mutations present at the time of AML diagnosis in the 295 patients in the cohort. Every patient is represented in an individual column, whereas genes and other AML features are listed in rows. Mutations are sorted by molecular ontogeny,4 with TP53 mutations listed first in green, secondary-type mutations (implying evolution from an antecedent MDS) in blue, and all other mutations (pan-AML/de novo type) below. NPM1 mutations and internal tandem duplications in FLT3 (FLT3-ITD) are in red, and DDX41 mutations are in gold. Complex cytogenetics are shown (top) above the TP53 mutations. The proportion of patients in the cohort with each alteration is reported on the right.
Mutations present at the time of diagnosis. Shown are all mutations present at the time of AML diagnosis in the 295 patients in the cohort. Every patient is represented in an individual column, whereas genes and other AML features are listed in rows. Mutations are sorted by molecular ontogeny,4 with TP53 mutations listed first in green, secondary-type mutations (implying evolution from an antecedent MDS) in blue, and all other mutations (pan-AML/de novo type) below. NPM1 mutations and internal tandem duplications in FLT3 (FLT3-ITD) are in red, and DDX41 mutations are in gold. Complex cytogenetics are shown (top) above the TP53 mutations. The proportion of patients in the cohort with each alteration is reported on the right.
Germline mutations that predispose to development of leukemia can present in older adults. We identified putative germline DDX41 mutations (median VAF = 0.48) resulting in start-loss (p.M1I) or premature termination codons in 16 of 295 patients (5.4%).17,18 Of those, 13 also had a second DDX41 mutation (median VAF = 0.09), most commonly affecting the somatic hotspot p.R525H.19 Five patients had germline rare variants in genes that regulate telomere maintenance (TERT [n = 2], TERC [n = 2], and RTEL1 [n = 1]), which have been linked to myeloid leukemogenesis in adults and NRM after transplantation.20,21 As expected, we did not identify likely pathogenic variants in genes associated with leukemia predisposition syndromes that typically presents earlier in life, such as SAMD9, SAMD9L, or SBDS.22-24
Clinical and genetic determinants of posttransplant LFS
To understand the relative impact of diagnostic and remission molecular status on posttransplant LFS in this cohort, we first developed an integrated model for LFS that included both genetic and nongenetic baseline characteristics. The overall workflow for the study is shown in Figure 2A. LFS and OS at 3 years after transplant for the full cohort were 42.1% and 46%, respectively, and the 3-year cumulative incidence of relapse was 38% (supplemental Figure 1). The 3-year incidence of NRM was 29%, which was higher than the observed NRM in the RIC arms of clinical trials in younger (CTN 0902; 4%)13 and older ( CTN 0502; 15%)8 patients with AML, but similar to NRM in retrospective studies of older patients who have undergone HCT (range, 30% to 40%).9,14,25-27
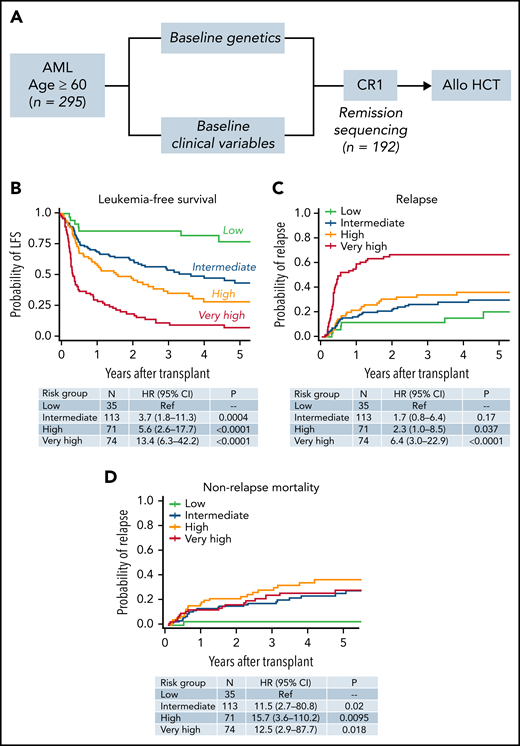
Development of an integrated model for LFS after transplantation. (A) The study workflow used to develop a prognostic model that included both genetic and nongenetic factors in the full cohort of 295 individuals. We additionally assessed whether the presence or absence of mutations at remission further refined the baseline mode in the 192 individuals with available remission samples. (B) LFS of individuals in the low (n = 35; green), intermediate (n = 113; blue), high (n = 71; orange), and very high (n = 77; red) risk groups. Cumulative incidence of relapse for the same groups (C), and the incidence of NRM (D). See supplemental Tables 8 to 10 for variables defining low, intermediate, high, and very high risk groups.
Development of an integrated model for LFS after transplantation. (A) The study workflow used to develop a prognostic model that included both genetic and nongenetic factors in the full cohort of 295 individuals. We additionally assessed whether the presence or absence of mutations at remission further refined the baseline mode in the 192 individuals with available remission samples. (B) LFS of individuals in the low (n = 35; green), intermediate (n = 113; blue), high (n = 71; orange), and very high (n = 77; red) risk groups. Cumulative incidence of relapse for the same groups (C), and the incidence of NRM (D). See supplemental Tables 8 to 10 for variables defining low, intermediate, high, and very high risk groups.
To identify gene mutations associated with LFS, we evaluated the 26 genes that were mutated in at least 10 patients (3%) in the study cohort. In univariable Cox models, mutations associated with inferior LFS included TP53 (HR 3.4; P < .001), JAK2 (HR 2.7; P < .001), FLT3-ITD without concomitant NPM1 mutation (HR 2.2; P < .001), and KRAS (HR 2.0; P = .004) (supplemental Table 6). The presence of an NPM1 mutation without concomitant FLT3-ITD was associated with prolonged LFS (HR 0.56, P = .002), as was the presence of a DDX41 mutation (HR 0.55; P = .036). We then generated a hierarchical model of molecular risk, in which unfavorable genetics included patients with TP53 or JAK2 mutations, or FLT3-ITDs without a concomitant NPM1 mutation (n = 63; 3-year LFS 6.7%); favorable genetics included patients without concomitant poor-risk mutations who had DDX41 or DNMT3A mutations, or NPM1 mutations without an FLT3-ITD (n = 95; 3-year LFS, 62%); intermediate genetics included all other patients (n = 137; 3-year LFS, 45%; P < .0001, for the 3-group comparison) (supplemental Figure 2).
Nongenetic factors related to the patient, disease, or transplant itself have been shown to influence LFS, NRM, and relapse.28,29 In univariable analysis, additional factors significantly associated with inferior LFS included pretransplant HCT comorbidity index (HCT-CI) ≥3 vs HCT-CI <3 (HR, 1.8; 95% confidence interval [CI], 1.3-2.7; P = .002), monosomal or nonmonosomal adverse karyotypes vs intermediate/favorable (HR, 4.4 for monosomal; 95% CI, 2.9-6.8; P < .001; HR 1.7 for adverse, nonmonosomal; 95% CI, 1.1-2.6; P = .013), CRi vs CR (HR, 1.8; 95% CI, 1.3-2.5; P < .001), and clinically defined secondary AML (sAML vs de novo AML; HR, 1.8; 95% CI, 1.3-2.4; P < .001) (supplemental Table 7).
To identify pretransplant factors that independently influence risk of death or relapse after transplantation in this cohort, we combined clinical and genetic variables in a multivariable frailty model adjusted for transplant center (Figure 2B). This model included the variables mentioned, as well as recipient age at the time of transplant, donor/recipient sex mismatch (male patient/female donor), intensive vs nonintensive induction therapy, white blood cell count at diagnosis, and receipt of consolidation therapy before transplant. Only molecular risk, karyotype, HCT-CI score, CRi, and clinically defined sAML retained significance and were included in the final model. Extended details are in supplemental Tables 8 to 13. Patients in the low-risk group (n = 35) had 3-year LFS of 86%, those in the intermediate-risk group (n = 113) had 3-year LFS 54% (vs low-risk; HR for death or relapse, 3.7; 95% CI, 1.8-11.3; P = .0004), those in the high-risk group (n = 71) had a 3-year LFS of 35% (HR, 5.6; 95% CI, 2.6-17.7; P < .0001), and those in the very high-risk group (n = 74) had a 3-year LFS of 9% (HR, 13.4; 95% CI, 6.3-42.2; P < .0001). The overall risk model reflects patients’ composite risk of relapse (Figure 2C) and NRM (Figure 2D).
Molecular genetics of CR
The presence of leukemia-associated gene mutations in CR has been associated with increased risk of relapse and inferior LFS after reduced-intensity allogeneic transplantation.13 To identify mutations present in remission at low abundance, we used a platform that incorporated duplex unique molecular identifiers, thereby enabling computational suppression of sequencing artifacts. By comparing remission with pretreatment time points, we defined each mutation as either persistent (pretreatment VAF ≥ 0.02 and detected in remission with ≥2 duplex read families) or emergent (pretreatment VAF = 0 or below 0.02 detection threshold and remission VAF ≥ 0.01) (supplemental Table 14). In total, we identified 352 persistent mutations, 326 of which (92.6%) met the VAF threshold of ≥0.001 proposed in recently published European LeukemiaNet guidelines for molecular MRD,30 and 100 emergent mutations.
Most patients (79.7%; 153 of 191 with diagnostic mutations) had at least 1 persistent mutation, and 150 of 153 had at least 1 mutation with VAF ≥ 0.001. The rate of persistence and the size of the persistent mutant clone varied by gene. Genes that are typically mutated in founding clones31,32 were more likely to have persistent mutations (Figures 3A; supplemental Figure 3; supplemental Table 15) present in high abundance (Figure 3B), including DNMT3A (55 of 66 persisted; 83.3%; median VAF = 0.064), TET2 (47 of 61; 77.0%; median VAF = 0.106), SF3B1 (6 of 7; 85.7%; median VAF = 0.025), U2AF1 (12 of 15; 80.0%; median VAF = 0.097), ASXL1 (30 of 38; 78.9%; median VAF = 0.084), and TP53 (20 of 26; 76.9%; median VAF = 0.044). In contrast, genes typically mutated in subclones were less likely to have persistent mutations, and mutations that persisted were present in low abundance, including CEBPA (0%), NPM1 (9.3%; median VAF = 0.003), WT1 (17.6%; median VAF = 0.003), FLT3-ITD (19.0%; median VAF = 0.004), FLT3-TKD/JMD (22.0%; median VAF = 0.001), KRAS (22.3%, median VAF = 0.022), and NRAS (25.0%; median VAF = 0.010). MRD assessed by multiparameter flow cytometry was available for 87 patients with remission molecular assessments; of 26 patients who were flow MRD positive, 21 (80.7%) were molecular MRD positive. Conversely, of 61 patients who were flow MRD negative, 32 (52.5%) were molecular MRD positive (supplemental Table 16).
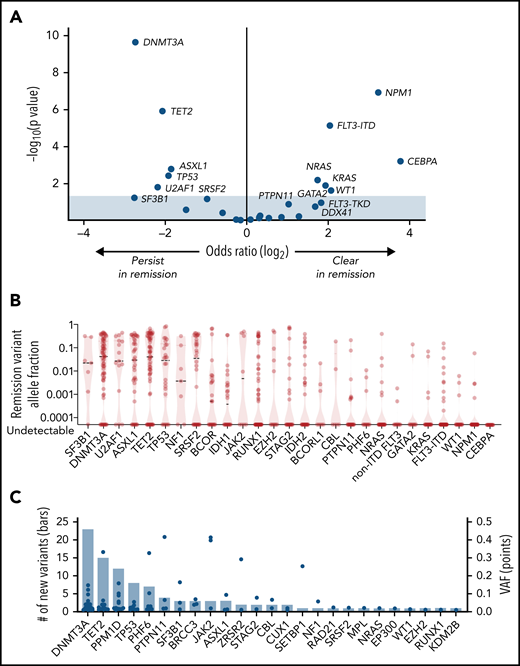
Characteristics of remission mutations. (A) Results of Fisher’s exact test for clearance or persistence of each gene, plotting the odds of clearance on the x-axis and significance, expressed as the negative log of the uncorrected P value, on the y-axis. Shown are mutations that are more likely to persist at remission (left) and those that are more likely to clear (right). (B) VAF of remission mutations. Median VAF is denoted by the dashed lines, and the range is indicated by the red violin plot for each gene. A version of this figure that excludes the 26 mutations with VAF < 0.001 is in supplemental Figure 3. (C) The number (left y-axis) and VAF (right y-axis) of mutations present at remission that were not detected at diagnosis, in descending order of frequency. VAFs are represented by blue dots and the total number of mutations is represented by the bars.
Characteristics of remission mutations. (A) Results of Fisher’s exact test for clearance or persistence of each gene, plotting the odds of clearance on the x-axis and significance, expressed as the negative log of the uncorrected P value, on the y-axis. Shown are mutations that are more likely to persist at remission (left) and those that are more likely to clear (right). (B) VAF of remission mutations. Median VAF is denoted by the dashed lines, and the range is indicated by the red violin plot for each gene. A version of this figure that excludes the 26 mutations with VAF < 0.001 is in supplemental Figure 3. (C) The number (left y-axis) and VAF (right y-axis) of mutations present at remission that were not detected at diagnosis, in descending order of frequency. VAFs are represented by blue dots and the total number of mutations is represented by the bars.
By comparing baseline and remission samples, we found that 66 of 192 patients (34.4%) had mutations that were newly detectable after treatment. The spectrum of emergent mutations was consistent with prior studies that reported clonal hematopoiesis after cytotoxic therapy33-35 and most commonly involved DNMT3A (23 mutations in 20 patients), TET2 (15 mutations in 13 patients), PPM1D (12 mutations in 10 patients), and TP53 (8 mutations in 8 patients) (Figure 3C).
Sole persisting DNMT3A, TET2, and ASXL1 mutations have been reported to have no impact on relapse risk and have been considered to reflect clonal hematopoiesis rather than frank residual leukemia.13,36 In older patients, however, ASXL1 mutations have been associated with evolution from prior MDS, whereas DNMT3A and TET2 mutations have not been.4,5 We found that DNMT3A and TET2 mutations commonly persisted in remission without other mutations (35 of 89 [39.3%] patients with DNMT3A or TET2 mutations at diagnosis remitted to only DNMT3A or TET2 mutations after treatment). In contrast, only 6 of 36 (16.7%) ASXL1 mutations persisted without other concomitant mutations, which were most commonly in MDS-associated genes such as SRSF2, RUNX1, and STAG2. Based on their different patterns of persistence and ontogenetic associations, we considered persistent sole DNMT3A or TET2 mutations as a group (DT), separate from the persistence of mutations in any other gene, including ASXL1 (MRD-positive).
MRD associations with baseline characteristics and posttransplant outcomes
The impact of molecular MRD on relapse risk after RIC HCT was defined based on the detection of remission mutations without knowledge of baseline genetic characteristics.13 Therefore, we sought to determine whether pretreatment factors were associated with the likelihood of MRD-positive CR. In multivariable logistic regression analysis, genetic ontogeny (secondary vs de novo ontogeny,4 odds ratio [OR], 7.9; 95% CI, 3.6-17; P < .0001; and TP53 vs de novo, OR, 7.7; 95% CI, 2.3-26; P = .001; and high-risk cytogenetics vs low risk OR, 5.7; 95% CI, 1.2-26.6; P = .027) were each associated with MRD positivity (Figure 4A; supplemental Table 17). For patients with MRD-positive remissions, remission clonal abundance, defined by the mutation with highest VAF per patient, was median VAF = 0.021 for patients with de novo genetic ontogeny, 0.042 for those with secondary ontogeny, and 0.113 for those with TP53 ontogeny (supplemental Figure 4). Most patients with DDX41 mutations at diagnosis had MRD-negative remissions.
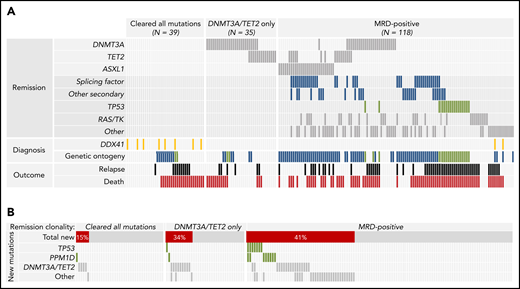
Clinical and genomic associations of molecular remission states. (A) Co-occurrence of nongenetic factors and specific persistent mutations for the 192 individuals in the remission cohort, grouped by type of molecular measurable residual disease (MRD). Patients who cleared all diagnostic mutations and patients with only persistent DNMT3A or TET2 mutations (together considered MRD negative) are in the left and middle respectively, and patients with other persistent mutations (MRD positive) are on the right. Remission DNMT3A, TET2, and ASXL1 mutations are shown at the top, with other classes of remission mutations shown next. Diagnostic genetic variables associated with molecular remission states are listed next, including DDX41 mutations (shown in gold) and molecular ontogeny (de novo in gray, secondary in blue, and TP53 in green). Patients who relapsed are shown in black and those who died are in red. (B) Distribution and co-occurrence of treatment-emergent mutations in patients with different patterns of molecular persistence. Patients who cleared all diagnostic mutations and patients with only persistent DNMT3A or TET2 mutations are in the left and middle, respectively, whereas patients with MRD-positive remissions are on the right.
Clinical and genomic associations of molecular remission states. (A) Co-occurrence of nongenetic factors and specific persistent mutations for the 192 individuals in the remission cohort, grouped by type of molecular measurable residual disease (MRD). Patients who cleared all diagnostic mutations and patients with only persistent DNMT3A or TET2 mutations (together considered MRD negative) are in the left and middle respectively, and patients with other persistent mutations (MRD positive) are on the right. Remission DNMT3A, TET2, and ASXL1 mutations are shown at the top, with other classes of remission mutations shown next. Diagnostic genetic variables associated with molecular remission states are listed next, including DDX41 mutations (shown in gold) and molecular ontogeny (de novo in gray, secondary in blue, and TP53 in green). Patients who relapsed are shown in black and those who died are in red. (B) Distribution and co-occurrence of treatment-emergent mutations in patients with different patterns of molecular persistence. Patients who cleared all diagnostic mutations and patients with only persistent DNMT3A or TET2 mutations are in the left and middle, respectively, whereas patients with MRD-positive remissions are on the right.
Posttreatment-emergent mutations may reflect either outgrowth of chemoresistant leukemic subclones or expansion of clonal hematopoiesis that is clonally unrelated to the leukemia itself.32,37 In patients who cleared diagnostic mutations or had only persistent DT mutations, treatment-emergent mutations were most commonly in DNMT3A and TET2 (13 of 20; 65%; Figure 4B). In contrast, the majority of newly detected TP53 and PPM1D mutations (15 of 18; 83%), as well as the majority of newly detected mutations in other genes (27 of 31; 87%) occurred in patients who also had persistent diagnostic mutations.
In univariable analysis, patients with MRD-positive remission had inferior LFS compared with those who had MRD-negative remission or had sole persistent DNMT3A/TET2 mutations (HR for death/relapse vs MRD negative/DT, 1.58; 95% CI, 1.07-2.32; P = .021; Figure 5A). These results were similar when reassigning 2 of 153 patients from MRDpositive to MRDnegative and 1 patient from DT to MRDnegative based on the provisional VAF threshold of 0.001 proposed in the European Leukemia Network guidelines for molecular MRD30 (supplemental Figure 5). The difference in LFS was driven by a higher rate of relapse in the MRD-positive group (at 3 years, 42.5% vs 20.6% for MRD negative/DT; P = .0006). The rates of relapse were unchanged when the definition of MRD negative was expanded to include sole persistent DNMT3A, TET2, or ASXL1 mutations (DTA; supplemental Figure 6). There was no significant difference in NRM based on MRD status (24.7% vs 17.4%, P = .24). When accounting for overall pretransplant risk, however, MRD positivity had no independent effect on LFS (Figure 5B). Three-year LFS was similar in patients with MRD-positive remission compared with MRD-negative/DT remission across risk groups: low risk (100% vs 86.7%; P = .39), intermediate risk (58.3% vs 53.6%; P = .75), high risk (37.7% vs 45.6%; P = .68), and very high risk (10.2% vs 12.5%; P = .93).
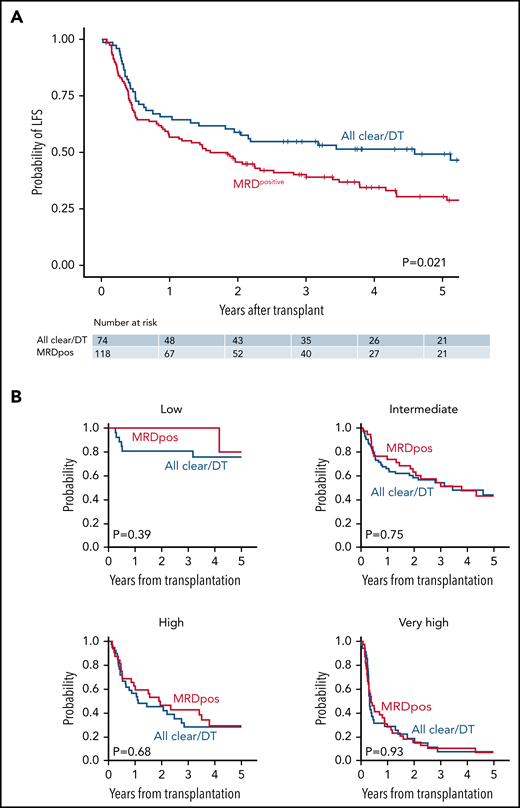
LFS according to baseline risk and MRD status. (A) Unadjusted LFS for all remission cohort patients (n = 192) according to MRD status. (B) LFS for remission cohort patients according to baseline risk status: low (n = 25); intermediate (n = 76); high (n = 40); or very high (n = 50). Patients with MRD-positive remissions are in red, and patients with MRD-negative remissions (all mutations cleared or DT only) are in blue.
LFS according to baseline risk and MRD status. (A) Unadjusted LFS for all remission cohort patients (n = 192) according to MRD status. (B) LFS for remission cohort patients according to baseline risk status: low (n = 25); intermediate (n = 76); high (n = 40); or very high (n = 50). Patients with MRD-positive remissions are in red, and patients with MRD-negative remissions (all mutations cleared or DT only) are in blue.
Discussion
AML in older adults has higher rates of induction failure and relapse than AML in younger adults.1,4,5,12 In this multi-institutional study of AML patients aged ≥60 undergoing allogeneic transplantation in first CR, we found that baseline clinical and genetic variables, but not remission MRD status, were the predominant determinants of posttransplant risks. High baseline molecular risk was driven independently by FLT3-ITD and TP53 mutations, which contributed to relapse risk, and by JAK2 mutations, which contributed to NRM, possibly as a consequence of potentiated inflammatory signaling that persisted after transplant.
We found that nearly 80% of patients had persistent mutations before transplant despite having achieved a morphologic CR. Whereas some patients had clonal remissions with only DNMT3A or TET2 mutations that did not impact outcomes, most had persistent MDS- and AML-associated mutations that were linked to an elevated risk of relapse. Recent results have indicated that intensification of transplant-preparative regimens could mitigate the risk of MRD-associated relapse.13 However, in this cohort of real-world older patients with AML, we observed a higher rate of NRM than was reported in clinical trials of older patients with AML8,38 or in younger patients who received RIC.13 These results raise the concern that intensified conditioning would not be tolerated in older patients with elevated vulnerability to treatment-related toxicity39 and highlights the need for novel approaches to mitigating risk of relapse in this population.
We found that the likelihood of MRD positivity was closely related to other baseline features of AML, including the presence of high-risk gene mutations, such as TP53, high-risk karyotypes, and prior MDS. Moreover, among patients with MRD-positive remissions, those with secondary genetic ontogeny or TP53 mutations at diagnosis had higher levels of molecular MRD than those with de novo genetic ontogeny. Previous studies have not determined whether AML mutations that are present at remission are an independent risk factor for relapse or reflect high-risk AML biology.13,36 Our findings suggest that the high rate of molecular persistence in this cohort may be related in particular to the evolution of many of these leukemias from MDS, consistent with the association between MDS and advancing age.
The distinction between molecular persistence and relapse risk is further supported by the observation that secondary genetic ontogeny was associated with persistence but not relapse, whereas clinically defined secondary AML was associated with relapse but not persistence. A possible contributor to this distinction is the higher rate of hypomethylating agents administered to patients with sAML before their AML diagnosis (40 of 91 patients with sAML [44%] vs 28 of 127 [22%] with secondary ontogeny), which may have selected for more therapy-resistant subclones that drove relapse in the sAML group. Consistent with this, the 3-year cumulative incidence of relapse (CIR) for sAML vs non-sAML was 45.8% vs 31.3% (P = .02), whereas the 3-year CIR for molecular ontogeny (de novo vs secondary vs TP53) was 28.3% vs 30.8% vs 84.8% (P < .0001).
In this cohort, MRD positivity did not have an independent impact on LFS after consideration of other pretransplant risk factors. Consistent with prior studies, MRD negativity was significantly associated with reduced risk of relapse, but this benefit was offset by elevated NRM among higher risk groups. These results are particularly important in the context of a recent study from a randomized cohort showing that MAC mitigates the relapse risk associated with MRD positivity.13 Although those findings have prompted a push to employ myeloablative regimens for eligible AML patients with persistent molecular disease after induction,40 they may not be applicable to many older patients, given the younger age of that cohort (median 55 and capped at 65), the small number of genes sequenced with omission of many MDS-associated genes commonly mutated in older patients with AML (such as SRSF2 and U2AF1), and the requirement that all patients be eligible for MAC before randomization. Indeed, although our cohort did not include enough patients receiving MAC to directly compare outcomes to RIC, the high rate of NRM in our and other older cohorts suggests in general that older patients with AML with MRD may not derive the same benefit from conventional treatment intensification as younger, fitter patients. Instead, older AML patients with persistent molecular disease before transplantation may benefit from nonintensive approaches aimed at achieving molecular clearance and reducing relapse risk, such as targeted MRD erasers after transplant maintenance therapy41-47 and, pending the results of ongoing clinical trials, novel monoclonal antibodies48 or immunologic therapies.49
Germline DDX41 mutations predispose to development of myeloid malignancies later in life,18 and were present in 5.4% of patients in this cohort. We found that DDX41 mutations were associated with overall favorable outcomes, including MRD-negative remission after initial therapy and prolonged LFS after HCT. The improved LFS in older patients with DDX41 mutations was driven entirely by a very low risk of relapse, and the risk of NRM was similar to that of other patients. This finding suggests that patients with germline DDX41 mutations may benefit from strategies to minimize transplant toxicity, including the selection of lower-intensity conditioning regimens associated with reduced risk of NRM50,51 or the use of posttransplant cyclophosphamide to minimize the risk of chronic graft-vs-host disease.52,53
Given the evolving nature of AML therapy, conclusions from retrospective analyses are fundamentally limited in their application to current and future therapies. Further, the time of MRD sample collection and assessment relative to initial induction, subsequent consolidation or maintenance therapies, and transplant-preparative regimen may contribute heterogeneity to observed outcome associations. As such, use of this baseline model to guide clinical decision making for older patients with AML will require validation by subsequent prospective studies of cohorts with uniform prior treatment histories and sample collection time points. Nevertheless, our results indicate that most of the posttransplant relapse risk in older patients with AML is encoded at the time of diagnosis and that the likelihood of molecular MRD positivity in these patients is primarily a reflection of baseline genetic risk. Attempting to eradicate persistent AML mutations with dose intensification may be counterproductive in older patients, given their high risk of treatment-related toxicity. This observation highlights both the importance of optimizing initial therapy for older patients with AML who are transplant candidates through use of liposomal daunorubicin/cytarabine and FLT3 inhibitors,54,55 as well as the development of nonintensive strategies for mitigating relapse risk in older patients who remain MRD positive before transplantation. Taken together, our results provide a framework for developing risk-adapted strategies and interpreting the relative prognostic impact of baseline and remission genetics in older patients with AML who are transplant candidates.
Acknowledgments
This work was supported by the ASH Medical Student Physician-Scientist and HONORS awards (H.M.M); National Institutes of Health, National Cancer Institute grants K08CA204734 (R.C.L.), K08CA263555 (C.J.G.), P01CA229092 (R.C.L., J.R., R.J.S), and PO1CA23766 and P30CA008748 (M.-A.P, C.C)]; the Frederick A. Deluca Foundation (R.C.L.); the James A. and Lois J. Champy Family Fund (R.C.L.); the Damon Runyon Cancer Research Foundation Physician Scientist Training Award (C.J.G.); The William G. Pomeroy Foundation Endowed Fund for AML Research (Dana-Farber Cancer Institute), and the Ted and Eileen Pasquarello Tissue Bank in Hematologic Malignancies (Dana-Farber Cancer Institute), and the Dana-Farber Cancer Institute Center for Cancer Genome Discovery (Anwesha Nag, Aaron Thorner, Audrey Dalgarno, and Neil Patel).
Authorship
Contribution: H.M.M., C.J.G., and R.C.L. designed the study, performed the data analysis, and wrote the manuscript; H.M.M. curated the sequencing results, clinical data, and variants; C.J.G. and H.K.T. performed the bioinformatic analysis and developed the data processing pipelines; H.T.K. performed statistical analyses and developed the prognostic models; B.H., P.V., N.D., B.S.M., S.G., and C.C. curated the clinical data; C.C., S.M., V.T.H., J.K., R.J.S., J.R. M.P.C., S.V., M.-A.P., E.S.W., L.P.G., S.D., and E.P.A. diagnosed the patients and prepared the samples; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: P.V. has received consulting fees from AbbVie, Amgen, Blueprint Medicines, CTI Biopharma, Incyte, Jazz Pharmaceuticals, Pfizer, and Novartis, and has served on the speakers bureau for Incyte. S.M has received clinical trial support from Gilead and Aprea. J.K. has served on the scientific advisory boards for Therakos, Cugene, and Biolojic Design and has received consulting fees from Amgen, Gentibio, Equillium, EMD Serono/Merck, and Moderna, as well as research support from BMS, Miltenyi Biotec, Novartis, Clinigen, and Regeneron. M.A.P reports honoraria from AbbVie, Astellas, Bristol-Myers Squibb, Celgene, Equilium, Incyte, Karyopharm, Kite/Gilead, Merck, Miltenyi Biotec, MorphoSys, Novartis, Nektar Therapeutics, Omeros, OrcaBio, Takeda, VectivBio AG, and Vor Biopharma; serves on the data and safety monitoring boards for Cidara Therapeutics, Medigene, Sellas Life Sciences, and Servier and the on the scientific advisory board of NexImmune; holds ownership interests in NexImmune and Omeros; and has received research support for clinical trials from Incyte, Kite/Gilead, Miltenyi Biotec, and Novartis. R.C.L. has received consulting fees from Takeda Pharmaceuticals and Bluebird Bio. E.S.W has received consulting honoraria from AbbVie, Astellas, BMS/Celgene, Genentech, GlaxoSmithKline, Jazz, Kite Pharmaceuticals, Kura Oncology, Novartis, Mana Therapeutics, Pfizer, Stemline Therapeutics, and Takeda; has served on the speakers’ bureau for Stemline Therapeutics, Kura, Pfizer, and DAVA Oncology; and is a member of the data and safety monitoring committees for AbbVie and Rafael Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: R. Coleman Lindsley, Dana-Farber Cancer Institute, 450 Brookline Ave – DA-530C, Boston, MA 02215; e-mail: coleman_lindsley@dfci.harvard.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
All genetic data required for replication are contained in the article and supplemental files.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal