

Key Points
TET2-mutant clonal hematopoiesis is associated with increased risk of incident gout.
Monosodium urate crystal treatment exacerbates Nlrp3-dependent IL-1β secretion and functional impairment in transplanted Tet2-knockout mice.

Abstract
Gout is a common inflammatory arthritis caused by precipitation of monosodium urate (MSU) crystals in individuals with hyperuricemia. Acute flares are accompanied by secretion of proinflammatory cytokines, including interleukin-1β (IL-1β). Clonal hematopoiesis of indeterminate potential (CHIP) is an age-related condition predisposing to hematologic cancers and cardiovascular disease. CHIP is associated with elevated IL-1β, thus we investigated CHIP as a risk factor for gout. To test the clinical association between CHIP and gout, we analyzed whole exome sequencing data from 177 824 individuals in the MGB Biobank (MGBB) and UK Biobank (UKB). In both cohorts, the frequency of gout was higher among individuals with CHIP than without CHIP (MGBB, CHIP with variant allele fraction [VAF] ≥2%: odds ratio [OR], 1.69; 95% CI, 1.09-2.61; P = .0189; UKB, CHIP with VAF ≥10%: OR, 1.25; 95% CI, 1.05-1.50; P = .0133). Moreover, individuals with CHIP and a VAF ≥10% had an increased risk of incident gout (UKB: hazard ratio [HR], 1.28; 95% CI, 1.06-1.55; P = .0107). In murine models of gout pathogenesis, animals with Tet2 knockout hematopoietic cells had exaggerated IL-1β secretion and paw edema upon administration of MSU crystals. Tet2 knockout macrophages elaborated higher levels of IL-1β in response to MSU crystals in vitro, which was ameliorated through genetic and pharmacologic Nlrp3 inflammasome inhibition. These studies show that TET2-mutant CHIP is associated with an increased risk of gout in humans and that MSU crystals lead to elevated IL-1β levels in Tet2 knockout murine models. We identify CHIP as an amplifier of NLRP3-dependent inflammatory responses to MSU crystals in patients with gout.
Introduction
Gout is the most common inflammatory arthritis in adults, leading to recurrent flares of debilitating joint pain and functional impairment.1-4 Gout is caused by precipitation of monosodium urate (MSU) crystals within and around joints of individuals with elevated serum urate levels (hyperuricemia).5-7 During an acute gout flare, MSU crystal deposits are phagocytized by macrophages, provoking an acute inflammatory response.8 This leads to activation of the NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome complex and release of the proinflammatory cytokine interleukin-1β (IL-1β), a key mediator of sustained inflammation in gout.8 Based on this rationale, clinical trials have evaluated IL-1β targeted agents for the treatment of gout and demonstrated therapeutic benefit.9,10
Several risk factors contribute to the development of gout, including male sex, obesity, age, chronic kidney disease (CKD), and high cell turnover states.3,4 Hyperuricemia is necessary but not sufficient for the development of gout, and factors that promote clinically evident gout in the presence of hyperuricemia remain poorly defined.
Clonal hematopoiesis of indeterminate potential (CHIP) is an age-associated precursor of myeloid malignancies.11,12 Individuals with CHIP have a clonal population of hematopoietic stem cells with somatic mutations in myeloid malignancy-associated genes at a variant allele frequency (VAF) ≥2% in the absence of a hematologic cancer.11,12 VAF is defined as the proportion of sequencing reads at a given genetic locus that bear the mutant allele and thus serves as a measure of clone size. Prior studies have demonstrated that individuals with a higher proportion of mutant cells in the peripheral blood (defined as VAF ≥10%) are at increased risk for developing not only hematologic malignancies but also nonmalignant disease phenotypes such as cardiovascular disease.13-16 Therefore, the VAF threshold ≥10% aims at distinguishing large mutant clones from smaller ones (defined as VAF <10%), which are less likely to be associated with inferior clinical outcomes. The epigenetic regulators DNMT3A and TET2 are the 2 most commonly mutated genes in CHIP.
Proinflammatory cytokine levels are aberrantly elevated in individuals with DNMT3A and TET2 mutations, suggesting a link among CHIP, systemic inflammation, and nonmalignant comorbidity.17 Activation of the NLRP3 inflammasome and release of inflammatory cytokines (eg, IL-1β) by Tet2-mutant immune cells have been identified as key drivers of aberrant inflammation in independent murine models.13,14 Enhanced NLRP3 inflammasome activity with secretion of IL-1β plays a key role in the pathophysiology of gout.8 We therefore evaluated whether CHIP may increase the risk of gout and whether Nlrp3 inflammasome inhibition may ameliorate the inflammatory response induced by MSU crystals.
Patients and methods
Study population
We examined the clinical association between CHIP and gout using 2 independent biorepositories. Whole exome sequencing (WES) data from peripheral blood samples obtained from the Mass General Brigham Biobank (MGBB, n = 8019) and the UK Biobank (UKB, n = 169 805; application ID 50834) were included in the analysis (supplemental Table 1). The MGBB comprises banked DNA samples from >80 000 adult individuals across the Mass General Brigham healthcare system in Boston, Massachusetts.18 The UKB is a prospective study that includes the genetic and clinical data of a cohort of 500 000 individuals aged 40 to 69 years who were enrolled between 2006 and 2010 from across the United Kingdom.19 Subjects with a diagnosis of any prevalent hematologic cancer prior to DNA collection were excluded. In total, 437 individuals (5.7% with CHIP) were diagnosed with gout before attending the UKB recruitment center, and 2567 individuals (8.4% with CHIP) had an incident gout diagnosis in the UKB. In the MGBB, we only assessed the total number of gout diagnoses (n = 203; 12.8% with CHIP) because of smaller sample size. The Kinship-Based Inference for Genome-Wide Association Studies tool was used to identify third-degree relatives who were not included in subsequent analyses.20 Among the related subjects, individuals with available WES or older participants were selected. Inclusion of individuals was not based on ancestry. Follow-up until March 2020 was included. Gout was defined through International Statistical Classification of Diseases and Related Health Problems (ICD) diagnostic codes with high specificity for identifying patients with gout and other crystal arthropathies (M10.00-M10.49, M11.00-M11.29, M11.80-11.99) without available information about longitudinal medication data or laboratory chemistry results.
WES and variant calling
WES data aligned to human genome reference (hg38) were obtained in cram format from the MGBB and UKB. Individuals with CHIP were identified based on a prespecified list of somatic variants in 71 genes recurrently mutated in myeloid cancers from WES using the Mutect2 algorithm as previously described (supplemental Table 2).13,21-23 The Genome Aggregation Database and a panel-of-normal exomes derived from WES of the youngest individuals (MGBB: n = 100; UKB: n = 500) were used to filter out sequencing artifacts and germline variants.24 The panel-of-normal exomes were manually curated to confirm that known CHIP variants were not present. Only variants meeting the following criteria were considered true somatic mutations: sequencing depth ≥20, variant reads ≥3, VAF ≥2%, and Genome Aggregation Database allele frequency <0.001. Any variants with a prevalence >1% in the analyzed cohort or VAF ≥35% were excluded unless previously reported to be somatic and involved in hematologic cancers. Insertions and deletions in homopolymer regions were included only if the number of reads supporting the alternate allele was ≥10 and VAF ≥10%.
Mouse models
The following mouse strains were used in this study: Tet2-floxed line B6;129S-Tet2tm1.1Iaai/J (JAX No. 017573), Dnmt3a-floxed line B6;129S4-Dnmt3atm3.1Enl/J (RIKEN BRC No. RBRC03731), and Nlrp3-floxed line B6;129S6-Nlrp3tm1Bhk/J (JAX No. 021302). Mice with constitutive expression of Cre recombinase under control of the Vav1 promoter were crossed with the Tet2-, Dnmt3a-, or Nlrp3-floxed line to generate animals with Tet2, Dnmt3a, and Nlrp3 knockout (KO) specific to the entire hematopoietic compartment. Wild-type Vav1-Cre animals were used as controls. For bone marrow transplantation, recipient CD45.1-postive mice were lethally irradiated and CD45.2-positive bone marrow was obtained from Tet2 or Dnmt3a KO sex- and age-matched littermates. Irradiation was split in 2 doses of 475 cGy within 3 hours. In total, 2 million donor cells were transplanted via retroorbital injection into each recipient animal. After 4 to 6 weeks of hematologic reconstitution, chimerism was checked by flow cytometry. Thereafter, mice were either administered intraperitoneal (peritonitis model, performed in Tet2 KO animals only) or subcutaneous (paw edema model, performed in Tet2 and Dnmt3a KO animals) injections of MSU crystals (Invivogen) to assess the inflammatory response.8,25 The murine models used in the present work had Tet2 or Dnmt3a KO, whereas CHIP in humans is a chimeric heterozygous state.
All experiments were performed with approval from the Institutional Animal Care and Use Committee (IACUC) of Brigham and Women’s Hospital and Dana-Farber Cancer Institute.
Peripheral blood analysis
Peripheral blood was obtained from the retroorbital sinus. Flow cytometry was performed using a BD FACSCanto II analyzer for chimerism assessment. After erythrocyte lysis, cells were resuspended in phosphate-buffered saline (PBS) supplemented with 2% fetal bovine serum and stained using the following antibody panel from BioLegend: CD45.1, CD45.2, CD3, CD11b, and B220.
Cell culture and macrophage stimulation
Whole bone marrow was obtained from murine long bones, hips, and spine. Bone marrow–derived macrophages (BMDM) were grown in Iscove's Modified Dulbecco's Medium with 10% fetal bovine serum, 1% penicillin/streptomycin/glutamine, and 10 ng/mL of murine macrophage colony-stimulating factor from animals with the following genotypes: wild-type (WT), Tet2 KO, Nlrp3 KO, Nlrp3 KO/Tet2 KO, and Dnmt3a KO/Nlrp3 KO. Macrophages were differentiated for 10 to 14 days and harvested thereafter using a cell scraper. BMDM were then replated into 24-well plates with 400 000 cells/well. After 24 hours, cells were stimulated with lipopolysaccharide (Invivogen) 1 ng/mL for 3 hours followed by administration of MSU crystals (Invivogen) for 6 hours. Controls received either PBS only or lipopolysaccharide followed by PBS.
RNA sequencing
Total RNA extraction was performed using the RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s instructions. Whole transcriptomic sequencing was carried out using Illumina NovaSeq 6000 (Novogene Corporation Inc.). For generation of sequencing libraries, the NEBNext Ultra RNALibrary Prep Kit for Illumina (NEB) was used following manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. Paired-end reads in FASTQ format were first processed through fastp.26 In this step, reads containing adapter and poly-N sequences and reads with low quality were removed. At the same time, Q20, Q30, and GC content of the clean data were calculated. All downstream analyses were based on the clean reads with high quality. The reads were then aligned to the mouse reference genome using the Spliced Transcripts Alignment to a Reference (STAR) software.27 FeatureCounts was used to count the reads mapped to each gene.28 Differential expression analysis between groups was performed using DESeq2 R package.29 The resulting P values were adjusted using the Benjamini and Hochberg approach for controlling the false discovery rate (FDR). Genes with an adjusted P < .05 found by DESeq2 were assigned as differentially expressed. Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis of differentially expressed genes was implemented by the clusterProfiler R package.30 Gene Ontology and KEGG terms with corrected P < .05 were considered significantly enriched by differential expressed genes.
Cytokine measurements
Cytokine analysis was performed by Eve Technologies Corporation using a multiplex immunoassay analyzed with a Bio-Plex 200 system. For mouse serum and tissue culture cytokine analysis, the MD44 array and MDHSCT18 array was used, respectively. All samples were handled according to the company’s instructions.
Histological analysis
Hind paws were fixed in 4% buffered formalin for 24 hours at room temperature and decalcified using Immunocal Decalcifier (StatLab) for 24 hours before paraffin embedding. After sectioning of paraffin-embedded tissue blocks, slides were stained using hematoxylin and eosin for evaluation of tissue inflammation by an anatomic pathologist.
Statistical analysis
Analyses were performed using R 3.4.4 or GraphpadPrism 9.3. Associations between CHIP and gout incidence were fitted using a Cox proportional hazards model adjusting for age (represented as deciles), sex (male or female), white blood cell count (WBC), CKD defined by estimated glomerular filtration rate (eGFR) and body mass index (BMI). Diagnosis of gout was defined as an event, and the censor date was the end of follow-up, the time of death, or the time of hematologic malignancy diagnosis, whichever came first. Associations with P < .05 were considered statistically significant.
Results
Clinical association between CHIP and gout
To investigate the association between CHIP and gout, we analyzed individual-level data from participants enrolled in the MGBB (n = 8019) and the UKB (n = 169 805) who had undergone exome sequencing from blood DNA. We hypothesized that the prevalence of CHIP at the time of blood draw for sequencing would be greater in patients with gout compared with those without. For the MGBB and the UKB studies, we identified n = 203 (2.5%) and n = 3004 (1.8%) individuals with a diagnosis for gout or other crystal arthropathies according to International Classification of Diseases (ICD) codes, respectively (supplemental Figure 1).
In total, we identified 645 CHIP variants in 590 (7.4%) individuals in the MGBB and 11 095 CHIP variants in 10 068 (5.9%) individuals in the UKB. Consistent with previous studies, the prevalence of CHIP increased with age. DNMT3A, TET2, and ASXL1 were the most frequently mutated genes, accounting for 78.8% and 83.6% of the cases in the MGBB and UKB, respectively. Most of the CHIP cases carried only 1 CHIP variant (ie, 91.7% in the MGBB and 91.2% in the UKB). Previously, large CHIP clones (VAF ≥10%) were strongly associated with clinical outcomes.11,13,16,17,31 In the MGBB and UKB, approximately half of the CHIP cases carried large CHIP clones (45.6% in the MGBB and 49.5% in the UKB).
Overall, we found gout to be more common in individuals with CHIP than without CHIP. In MGBB, 26 of 590 cases with CHIP and VAF ≥2% had gout (4.4%) vs 177 of 7429 controls (2.4%) without CHIP (OR, 1.69; 95% CI, 1.09-2.61; P < .05) (Figure 1A). In UKB, 141 of 5000 cases with CHIP and VAF ≥10% had gout (2.8%) vs 2763 of 159 737 controls (1.7%) without CHIP (OR, 1.25; 95% CI, 1.05-1.50; P < .05 (Figure 1B). Subgroup analyses within the UKB study revealed no significant difference in gout prevalence between CHIP cases with VAF <10% and cases without CHIP (2.0% vs 1.7%; OR, 0.93; 95% CI, 0.76-1.15; P = .50) (Figure 1B).
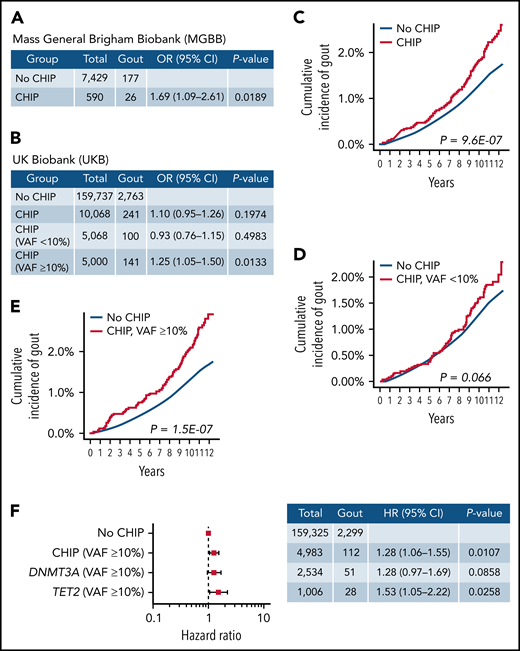
CHIP is associated with increased risk of gout. (A) Association between CHIP and gout in the MGBB. Generalized linear model adjusting for age deciles and sex was used to calculate OR and 95% CI. Individuals without CHIP were used as reference group. (B) Association between CHIP and gout in the UKB. Generalized linear model adjusting for age deciles, sex, WBC, eGFR, and BMI was used to calculate OR and 95% CI. Individuals without CHIP were used as reference group. (C) Cumulative incidence of gout in individuals with and without CHIP in UKB. Individuals were identified as events at the time of gout diagnosis and censored at the end of follow-up, at the time of death, or at the time of hematologic malignancy diagnosis. (D) Cumulative incidence of gout among individuals without CHIP and CHIP with VAF <10% in UKB. Individuals were identified as events at the time of gout diagnosis and censored at the end of follow-up, at the time of death, or at the time of hematologic malignancy diagnosis. (E) Cumulative incidence of gout among individuals without CHIP and CHIP with VAF ≥10% in UKB. Individuals were identified as events at the time of gout diagnosis and censored at the end of follow-up, at the time of death, or at the time of hematologic malignancy diagnosis. (F) Forest plot of HR for the association between CHIP and gout in the UKB. Cox proportional hazards model adjusting for age deciles, sex, WBC, eGFR, and BMI was used to calculate HR and 95% CI. Individuals without CHIP were used as reference group.
CHIP is associated with increased risk of gout. (A) Association between CHIP and gout in the MGBB. Generalized linear model adjusting for age deciles and sex was used to calculate OR and 95% CI. Individuals without CHIP were used as reference group. (B) Association between CHIP and gout in the UKB. Generalized linear model adjusting for age deciles, sex, WBC, eGFR, and BMI was used to calculate OR and 95% CI. Individuals without CHIP were used as reference group. (C) Cumulative incidence of gout in individuals with and without CHIP in UKB. Individuals were identified as events at the time of gout diagnosis and censored at the end of follow-up, at the time of death, or at the time of hematologic malignancy diagnosis. (D) Cumulative incidence of gout among individuals without CHIP and CHIP with VAF <10% in UKB. Individuals were identified as events at the time of gout diagnosis and censored at the end of follow-up, at the time of death, or at the time of hematologic malignancy diagnosis. (E) Cumulative incidence of gout among individuals without CHIP and CHIP with VAF ≥10% in UKB. Individuals were identified as events at the time of gout diagnosis and censored at the end of follow-up, at the time of death, or at the time of hematologic malignancy diagnosis. (F) Forest plot of HR for the association between CHIP and gout in the UKB. Cox proportional hazards model adjusting for age deciles, sex, WBC, eGFR, and BMI was used to calculate HR and 95% CI. Individuals without CHIP were used as reference group.
Given the increased frequency of gout among CHIP carriers, we next assessed the incidence of gout in cases with and without CHIP. Because of the significantly smaller sample size and lack of longitudinal follow-up within MGBB, the incidence analyses were restricted to UKB. In a univariate analysis, CHIP carriers were associated with an increased incidence of gout compared with individuals without CHIP (HR, 1.43; 95% CI, 1.24-1.66; P < .0001) (Figure 1C). There was no significant difference in incident gout between carriers of small clones (VAF <10%) and individuals without CHIP (HR, 1.22; 95% CI, 0.99-1.51; P = .066) (Figure 1D). However, individuals with large clones (VAF ≥10%) were associated with higher gout incidence compared with individuals without CHIP (HR, 1.66; 95% CI, 1.37-2.00; P < .0001) (Figure 1E). Using a Cox proportional hazards model, CHIP with VAF ≥10% was associated with 1.28 times the risk of incident gout compared with no CHIP after adjusting for common gout risk factors (age, sex, WBC, CKD, and BMI) (HR, 1.28; 95% CI, 1.06-1.55; P < .05) (Figure 1F). Gene-specific analyses revealed that large TET2 clones with VAF ≥10% were independently associated with 1.53 times the risk of incident gout (HR, 1.53; 95% CI, 1.05-2.22; P < .05) (Figure 1F). We also assessed the association between CHIP and gout in male and female patients separately. Although this analysis was underpowered because of small sample sizes, comparable effects of CHIP on incident gout were identified in both sexes. We observed a trend toward increased risk of gout in individuals with large CHIP clones (VAF ≥10%) in both males and females.
We next asked whether the presence of CHIP increased the risk of gout in individuals with hyperuricemia. Serum urate levels were strongly associated with gout both in females and males: individuals with serum urate levels above the median were more likely to develop gout than those with serum levels below the median (Figure 2A). In an analysis of incident gout disease in individuals with hyperuricemia (defined as serum urate levels above the median), CHIP with VAF ≥10% was independently associated with a 1.27-fold risk of incident gout after adjustment for age, sex, WBC, CKD, and BMI (HR, 1.27; 95% CI, 1.02-1.58; P < .05) (Figure 2B). The presence of a TET2 clone with VAF ≥10% was associated with a 1.60-fold risk of incident gout in individuals with hyperuricemia (HR, 1.60; 95% CI, 1.04-2.47; P < .05) (Figure 2B).
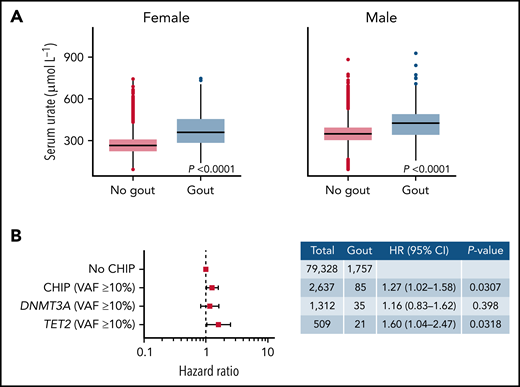
CHIP increases risk of gout in the presence of elevated serum urate levels. (A) Association between serum urate levels and gout diagnosis in males and females. P values were computed using Wilcoxon rank-sum test. (B) Forest plot of HR for the association between CHIP and gout. This analysis included individuals with serum urate levels above the median of UKB. Cox proportional hazards model adjusting for age deciles, sex, WBC, eGFR, and BMI was used to calculate HR and 95% confidence intervals (95% CI). Individuals without CHIP were used as reference group.
CHIP increases risk of gout in the presence of elevated serum urate levels. (A) Association between serum urate levels and gout diagnosis in males and females. P values were computed using Wilcoxon rank-sum test. (B) Forest plot of HR for the association between CHIP and gout. This analysis included individuals with serum urate levels above the median of UKB. Cox proportional hazards model adjusting for age deciles, sex, WBC, eGFR, and BMI was used to calculate HR and 95% confidence intervals (95% CI). Individuals without CHIP were used as reference group.
Administration of MSU crystals increases IL-1β secretion and paw edema in Tet2-deficient mice
Based on the human observation that TET2 mutations were associated with gout, we examined whether hematopoietic-specific Tet2 inactivation could causally influence the gout phenotype in a murine model. Previous studies demonstrated that intraperitoneal injection of MSU crystals in mice leads to recruitment of myeloid cells and inflammasome activation with release of IL-1β.8 We examined whether mice with Tet2 (KO) hematopoietic cells have altered inflammation in response to MSU crystals. We transplanted bone marrow from WT (Vav1-Cre) and Tet2-deficient (Tet2fl/fl; Vav1-Cre) animals into lethally irradiated mice. After hematopoietic recovery (4-6 weeks), WT and Tet2 KO mice were injected with MSU crystals intraperitoneally. Following treatment with MSU crystals, transplanted Tet2 KO animals had increased serum levels of IL-1β, C-C chemokine ligand 22 (CCL22), and C-C chemokine ligand 5 (CCL5) as compared with WT (P < .05 and FDR < 0.1) (Figure 3A). These cytokines have been reported to elicit highly proinflammatory effects through leukocyte recruitment and activation of the innate immune response. Using flow cytometry, we found a similar increase of myeloid (CD11b+) cells with concomitant decrease in B cells (B220+) in both peripheral blood and peritoneal fluid after administration of MSU crystals compared with PBS among WT and transplanted Tet2 KO animals (Figure 3B). Consistent with prior studies, these results confirm that intraperitoneal injection of MSU leads to an acute inflammatory response and influx of myeloid cells after 6 hours.8 However, we did not observe any statistical difference in proportion of immune cells, specifically CD11b+ cells, between Tet2 KO and WT animals treated with MSU crystals.
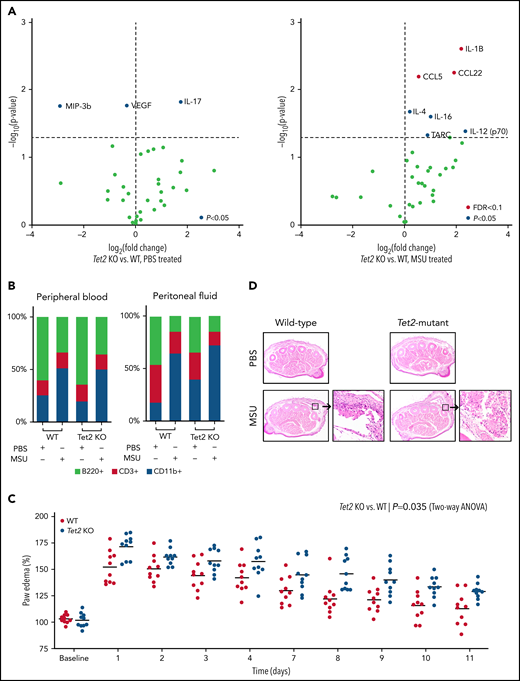
Loss of Tet2 exacerbates MSU crystal-induced inflammatory phenotype in murine models. (A) Serum cytokine array of Tet2-deficient (Tet2 KO, n = 5) vs WT (n = 5) mice after 6 hours of treatment with PBS (left plot) and MSU crystals (right plot). Blue dots highlight cytokines with P < .05 using 2-sample Student t test; red dots highlight cytokines with adjustment of false discovery rate (FDR < 0.1). (B) Flow cytometry analysis of peripheral blood and peritoneal fluid from WT and Tet2 KO mice after 6 hours of treatment with PBS and MSU crystals. Two-sample Student t test was used to compare the fraction of CD11b+ cells between MSU-treated WT and Tet2 KO animals. (C) Paw edema elicited by subcutaneous injection of MSU crystals into the foot pads of WT (n = 10) and Tet2 KO (n = 10) mice. P values were calculated using 2-way analysis of variance. (D) Representative hematoxylin and eosin stained paw cross sections of WT and Tet2 KO mice treated with PBS and MSU crystals shows increased cellular infiltrate (40× magnification).
Loss of Tet2 exacerbates MSU crystal-induced inflammatory phenotype in murine models. (A) Serum cytokine array of Tet2-deficient (Tet2 KO, n = 5) vs WT (n = 5) mice after 6 hours of treatment with PBS (left plot) and MSU crystals (right plot). Blue dots highlight cytokines with P < .05 using 2-sample Student t test; red dots highlight cytokines with adjustment of false discovery rate (FDR < 0.1). (B) Flow cytometry analysis of peripheral blood and peritoneal fluid from WT and Tet2 KO mice after 6 hours of treatment with PBS and MSU crystals. Two-sample Student t test was used to compare the fraction of CD11b+ cells between MSU-treated WT and Tet2 KO animals. (C) Paw edema elicited by subcutaneous injection of MSU crystals into the foot pads of WT (n = 10) and Tet2 KO (n = 10) mice. P values were calculated using 2-way analysis of variance. (D) Representative hematoxylin and eosin stained paw cross sections of WT and Tet2 KO mice treated with PBS and MSU crystals shows increased cellular infiltrate (40× magnification).
Having demonstrated that MSU crystal-mediated peritonitis results in elevated inflammatory cytokines in mice transplanted with Tet2 KO hematopoietic cells, we used a second model that more closely reflects the clinical phenotype of gout and examined edema formation after MSU injection into the paw.25 In this model, paw edema was defined as the relative thickness of the foot injected with MSU crystals (left hind paw) normalized to the contralateral PBS-injected foot (right hind paw). Mice received a single subcutaneous injection of MSU crystals and PBS into the footpad. Following treatment with MSU crystals, transplanted Tet2 KO animals had exacerbated paw swelling compared with WT mice over an extended period of 2 weeks (P < .05) (Figure 3C). Histologic examination confirmed the increased inflammation in mice with transplanted Tet2 KO hematopoietic cells, as evidenced by marked edema formation and macrophage-predominant inflammatory infiltrates, compared with transplanted WT mice (Figure 3D). Our in vivo experiments demonstrate both increased IL-1β cytokine secretion and higher degree of functional impairment in MSU-treated animals transplanted with Tet2 KO compared with WT cells. We did not observe enhanced paw edema in transplanted Dnmt3a-deficient mice when compared with WT animals (supplemental Figure 3).
Tet2-mutant macrophages secrete higher levels of IL-1β in the presence of MSU
Given that proinflammatory cytokines correlated with an increased fraction of myeloid cells in the peripheral blood of mice, we next examined whether isolated macrophages from Tet2 KO mice release altered levels of inflammatory cytokines. BMDM from Tet2 KO mice were generated to investigate the MSU crystal-induced cytokine profile in mutant macrophages. Following treatment with MSU crystals, IL-1β secretion was significantly higher in Tet2 KO macrophages across a broad range of time points (2 hours: P < .01; 3 hours: P < .001; 6 hours: P < .001; 12 hours: P < .0001; 18 hours: P < .001; 24 hours: P < .01; 48 hours: P < .05) (Figure 4A). In accordance with the results from our in vivo experiments, IL-1β was found to be the highest differently secreted cytokine in both Tet2-deficient BMDM compared with WT cells (P < .05 and FDR < 0.1) (Figure 4B). IL-1β levels did not differ between mutant and WT BMDM at baseline when treated with PBS (supplemental Figure 2A-B). In line with our results from Tet2 KO BMDM, we found increased IL-1β secretion in Dnmt3a KO BMDM when compared with WT (P < .05 and FDR < 0.1) (supplemental Figure 4A).
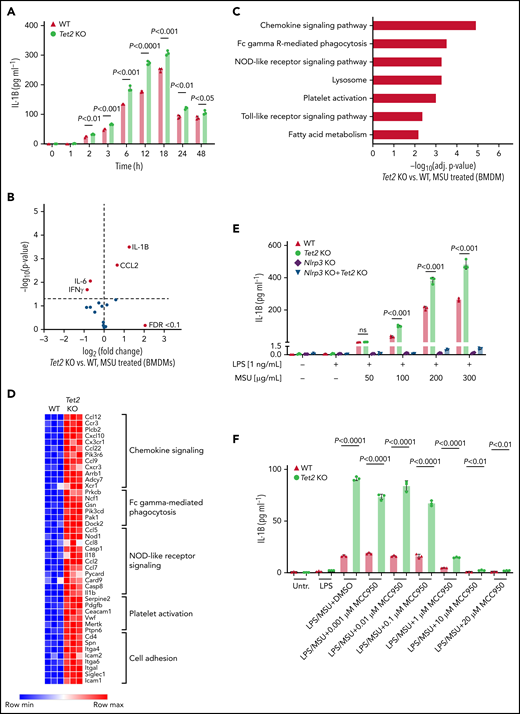
Loss of Tet2 augments MSU crystal-induced secretion of IL-1β in macrophages. (A) Time-course analysis of IL-1β levels in supernatant of WT and Tet2 KO BMDMs treated with an MSU crystal dose of 100 µg/mL. P values were obtained using 2-sample Student t test. (B) Cytokine array in supernatant of Tet2 KO vs WT BMDMs after 6 hours of treatment with MSU crystals. Red dots highlight cytokines with FDR < 0.1. (C) KEGG pathway enrichment analysis of differentially expressed genes in Tet2 KO vs WT BMDMs treated with MSU crystals. (D) Heatmap of selected differentially expressed genes in inflammatory pathways from Tet2 KO vs WT BMDMs after administration of MSU crystals. (E) Dose-response analysis of IL-1β levels in supernatant of MSU crystal-treated BMDMs obtained from WT, Tet2 KO, Nlrp3 KO, and Tet2 KO+Nlrp3 KO mice. P values were obtained using 2-sample Student t test. (F) Dose-response analysis of IL-1β levels in supernatant of MSU crystal-treated BMDMs incubated with MCC950. BMDMs were obtained from WT and Tet2 KO mice. P values were obtained using 2-sample Student t test.
Loss of Tet2 augments MSU crystal-induced secretion of IL-1β in macrophages. (A) Time-course analysis of IL-1β levels in supernatant of WT and Tet2 KO BMDMs treated with an MSU crystal dose of 100 µg/mL. P values were obtained using 2-sample Student t test. (B) Cytokine array in supernatant of Tet2 KO vs WT BMDMs after 6 hours of treatment with MSU crystals. Red dots highlight cytokines with FDR < 0.1. (C) KEGG pathway enrichment analysis of differentially expressed genes in Tet2 KO vs WT BMDMs treated with MSU crystals. (D) Heatmap of selected differentially expressed genes in inflammatory pathways from Tet2 KO vs WT BMDMs after administration of MSU crystals. (E) Dose-response analysis of IL-1β levels in supernatant of MSU crystal-treated BMDMs obtained from WT, Tet2 KO, Nlrp3 KO, and Tet2 KO+Nlrp3 KO mice. P values were obtained using 2-sample Student t test. (F) Dose-response analysis of IL-1β levels in supernatant of MSU crystal-treated BMDMs incubated with MCC950. BMDMs were obtained from WT and Tet2 KO mice. P values were obtained using 2-sample Student t test.
To characterize the full range of inflammatory response of MSU-treated macrophages, we performed mRNA sequencing in Tet2 KO and WT cells. KEGG enrichment analysis of pathways differentially expressed after Tet2 loss in macrophages showed strong signatures of chemokine signaling, phagocytosis, and NOD-like receptor (NLR) signaling (Figure 4C-D).
Inflammasome inhibition abrogates IL-1β secretion in Tet2-deficient macrophages
Our data implicate inflammasome activation and IL-1β secretion in the biology of Tet2-mediated response to MSU crystals, highlighting an opportunity for therapeutic strategies using inflammasome inhibitors. To determine whether inflammasome blockade can reduce CHIP-mediated inflammation, BMDM from Tet2 KO, Dnmt3a KO, Tet2 KO+Nlrp3 KO, Dnmt3a KO+Nlrp3 KO, and WT animals was treated with increasing doses of MSU. While Tet2 KO cells showed augmented IL-1β secretion as compared with WT, IL-1β release was abrogated in macrophages with genetic inflammasome inhibition even in the presence of Tet2 KO (Figure 4E). We then tested whether therapeutic Nlrp3 inflammasome inhibition using MCC950 (Invivogen) would ameliorate the phenotype and demonstrate markedly reduced IL-1β cytokine levels in MSU-treated macrophages with and without Tet2 loss (Figure 4F). Comparable results were obtained for Dnmt3a KO macrophages in which IL-1β secretion was Nlrp3-dependent (supplemental Figure 4B).
Discussion
CHIP is a precursor of myeloid malignancies and has been implicated in the pathogenesis of atherosclerosis.13,14 More recently, the contribution of CHIP to the pathogenesis of other systemic inflammatory conditions (ie, hemophagocytic lymphohistiocytosis, osteoporosis, and chronic obstructive pulmonary disease) has been identified.15,16,31 Here, we establish a clinical and biological association between CHIP and gout, a highly prevalent arthritis. In human genetic studies from 2 independent cohorts, we found a strong association between CHIP and gout, particularly in individuals with large clones (VAF ≥10%). Experimental studies demonstrate a causal relationship between Tet2 inactivation and gout, with higher degrees of functional impairment in Tet2-deficient mouse models compared with WT. Cytokine analysis indicated that Tet2 inactivation exaggerates the gout phenotype via a mechanism that includes enhanced IL-1β secretion in macrophages in response to MSU crystals. One limitation of this study is the use of single serum urate levels due to missing longitudinal measurements, which may not fully reflect the nature of chronic exposure to hyperuricemia in patients with gout. However, our analysis of the UKB data demonstrated an association between elevated serum urate levels and gout.
Prior studies have investigated the role of the NLRP3 inflammasome in gout pathophysiology, but it has remained unexplained why a subset of patients with gout have a much higher degree of inflammation than others. CHIP has been identified as an amplifier of inflammation through aberrant immune activation and IL-1β secretion.13,14,21,32,33 While aberrant IL-1β secretion has previously been identified in CHIP-positive cardiovascular disease,13,14 here we demonstrate a novel role for the NLRP3 inflammasome in CHIP-associated gouty arthritis. Our findings indicate that Tet2 CHIP results in higher dose-dependent increase of IL-1β cytokine levels and macrophage activation. Taken together, these findings suggest that CHIP exaggerates the inflammatory response in individuals with gout.
Administration of the anti–IL-1β antibody canakinumab has been associated with reduced risk of gout.34 Our studies highlight IL-1β as a potential therapeutic target and provide the biologic rationale for evaluating therapeutic IL-1β inhibition in patients with CHIP-positive gout. We found that both genetic and pharmacologic inflammasome inhibition abrogates IL-1β secretion in Tet2 and Dnmt3a knockout macrophages in the presence of MSU and thereby provide a proof of concept for efficacy of inflammasome inhibition in patients with CHIP-positive gout. More broadly, our findings substantiate CHIP as a biomarker for certain types of inflammatory disorders, and screening for CHIP may identify a subset of individuals most likely to benefit from IL-1β blockade or NLRP3 inflammasome inhibition to prevent or treat nonmalignant inflammatory diseases.
In summary, this work establishes TET2-mutant CHIP as a modifier of gout in humans. TET2-mutant CHIP is associated with an increased risk of having and developing gout in human cohorts, and mouse models confirm a direct influence of Tet2 KO hematopoietic cells on gout-induced inflammation and arthropathy. Hyperuricemia is common in patients with myeloid malignancies as a result of high cell turnover states. The aberrant inflammatory response to hyperuricemia in such patients may be related to mutant macrophages. CHIP may provide a mechanistic explanation for the heterogeneity in clinical symptoms and inflammation due to gout. Our findings substantiate the biologic rationale for future interventional strategies directed at CHIP-associated gout and other inflammatory conditions.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, AG252/1-1) (M.A.); funds from the Knut and Alice Wallenberg Foundation (KAW2017.0436) (A.N.); the Arthritis National Research Foundation (P.C.); the ASH/RWJF Harold Amos Medical Faculty Development Program (L.D.W.); the National Cancer Institute, National Institutes of Health (K08CA241085) and the Young Investigator Award of the Sarcoma Alliance for Research through Collaboration (SARC) (I.-M.S.); grants from the National Heart, Lung, and Blood Institute, National Institutes of Health (R01HL148050, R01HL151283, R01HL148565), the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (R01DK125782), Fondation Leducq (TNE-18CVD04), and Massachusetts General Hospital (Fireman Chair) (P.N.); the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (P30AR070253) (P.A.N.); a Career Award for Medical Scientists from the Burroughs Wellcome Fund and the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health (K08AR072791, P30AR070253) (D.A.R.); and the National Heart, Lung, and Blood Institute and the National Cancer Institute, National Institutes of Health (R01HL082945, P01CA066996, P50CA206963, and R35CA253125), the Howard Hughes Medical Institute, and the Adelson Medical Research Foundation (B.L.E.).
Authorship
Contribution: M.A. and B.L.E. designed and initiated the project; M.A. designed, performed, and analyzed experiments; A.N., L.D.W., C.J.G., A.S., A.G.B., M.M.U., G.K.G., S.J., and P.N. generated somatic mutation calls for UK Biobank and Mass General Brigham Biobank studies; A.N. and D.N. performed statistical analyses and interpreted the data; M.A., P.C., M.M., V.S., P.G.K., W.J.W., A.E.L., P.G.M., I.-M.S., P.A.N., and D.A.R. performed in vivo and in vitro experiments; M.A. and B.L.E. drafted the manuscript; all authors made substantial contributions to data analysis and/or interpretation, drafted and/or revised the manuscript, and approved the final version for publication.
Conflict-of-interest disclosure: M.A. received consulting fees from German Accelerator Life Sciences and is co-founder and shareholder of iuvando Health, all unrelated to the present work. P.G.M. has received consulting fees from Foundation Medicine in the past 3 years. R.M.S. has received grant support from AbbVie, Agios, Arog, and Novartis, personal fees from AbbVie, AMGEN, Aprea, Actinium, Agios, Argenx, Astellas, AstraZeneca, BerGenBio, Biolinerx, Bristol-Myers Squibb, Celgene, Daiichi-Sankyo, Elevate, Foghorn Therapeutics, Gemoab, GlaxoSmithKline, Innate, Janssen, Jazz, Macrogenics, Novartis, OncoNova, Otsuka, Pfizer, Roche, Stemline, Syndax, Syntrix, Syros, Takeda, and Trovagene outside the submitted work. P.N. has received investigator-initiated grants from Amgen, Apple, AstraZeneca, Boston Scientific, and Novartis; personal fees from Apple, AstraZeneca, Blackstone Life Sciences, Foresite Labs, Novartis, and Roche/Genentech; is a co-founder of TenSixteen Bio; is a shareholder of geneXwell and TenSixteen Bio; and reports spousal employment at Vertex, all unrelated to the present work. D.A.R. received grant support from Janssen and Bristol-Myers Squibb and personal fees from Pfizer, Janssen, Merck, Scipher Medicine, GlaxoSmithKline, and Bristol-Myers Squibb outside the submitted work. B.L.E. received research funding from Celgene, Deerfield, Novartis, and Calico and consulting fees from GRAIL and is a member of the scientific advisory board and is shareholder for Neomorph Therapeutics, TenSixteen Bio, Skyhawk Therapeutics, and Exo Therapeutics.
Correspondence: Benjamin L. Ebert, Dana-Farber Cancer Institute, Department of Medical Oncology, D1610A, 450 Brookline Ave, Boston, MA 02115; e-mail: Benjamin_Ebert@DFCI.Harvard.edu.
Data is available upon request by emailing corresponding author.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal