

TO THE EDITOR:
Although severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections have spread worldwide, vaccines are highly effective in preventing severe COVID-19 and mortality.1 The emergence of the Omicron variant with more than 30 mutations in the spike protein drastically reduced the neutralizing ability of vaccine-induced antibodies,2 leading to frequent breakthrough infections.3 Nevertheless, vaccine-induced immunity remains highly effective in preventing severe COVID-19.4 This is probably mediated by Spike-specific T cells, whose inductions have been shown to correlate with onset of vaccine efficacy5 and are largely unaffected by Omicron mutations.6-9
CD19-targeting chimeric antigen receptor (CAR) T-cell therapy is a major breakthrough in the treatment of relapsed/refractory B-lineage malignancies, whose bystander effect is long-term B-cell aplasia.10 Consequently, these patients do not mount a humoral response after COVID-19 vaccination.11-13 Initial data demonstrated an induction of Spike-specific CD4 T cells in adult patients14; however, an in-depth characterization of vaccine-induced T-cell responses and their resistance to emerging viral variants is lacking.
Here, we longitudinally studied anti-CD19 4-1BB-CD3z-CAR T cell–treated patients, mostly adolescents and young adults, before and after the first and second dose of vaccination with BNT162b2. We analyzed the magnitude of the T-cell response, the phenotypes of this response, their multispecificity, and their ability to respond to SARS-CoV-2 variants B.1.617.2 (Delta) and B.1.1.529 (Omicron).
We recruited 8 patients who had received single infusions of anti-CD19 CAR T cells at least 6 months before 2-dose vaccination with BNT162b2 3 weeks apart (supplemental Table 1 available on the Blood Web site) and 26 healthy controls (supplemental Table 2). None of the individuals in the study were infected with SARS-CoV-2 before vaccination or during the study period; none of the patients were receiving any immunosuppressive ,therapies and all were clinically well. Other baseline and disease specific characteristics are described in supplemental Table 1.
All 7 adolescents and young adults, except the 1 elderly patient in the study, experienced mild adverse events (local and systemic) especially after the second dose (Figure 1A). Seven of 8 patients mounted none to only minimal levels of Spike-specific antibodies following vaccination (Figure 1B). Only patient 9, who lost persistence of CAR T cells at the beginning of the study period and subsequently had detectable B cells, demonstrated an antibody response after the second dose.
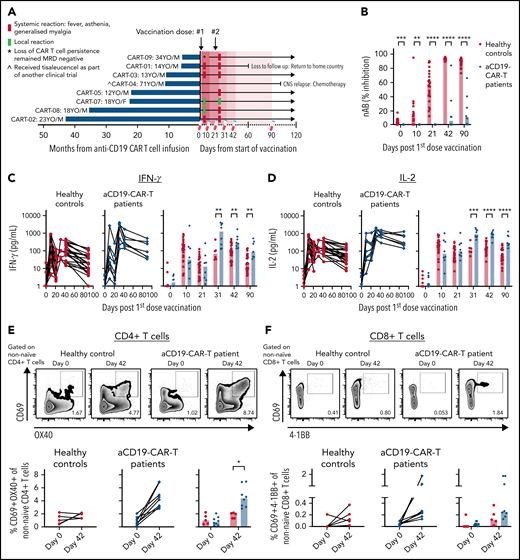
Enhanced vaccine-induced Spike-specific T-cell response in anti-CD19 CAR T cell–treated patients. Healthy individuals (n = 26) and (A) anti-CD19 CAR T cell–treated patients (n = 8) were vaccinated on days 0 and 21 with BNT162b2 mRNA vaccine. Blood samples were taken on days 0, 11, 21, 31, 42, and 90. (B) Levels of neutralizing antibodies. IFN-γ (C) and IL-2 (D) secreted into the plasma after whole blood stimulation with Spike-peptide pool and dimethyl sulfoxide control were quantified. (E) Representative flow cytometry plots for quantifying AIM+CD4 T cells. The numbers represent the percentage of total nonnaïve CD4 T cells that are AIM+ on days 0 and 42. Below, summary data of AIM+CD4 T-cell frequency before and after vaccination. The values represent the background-subtracted frequency of AIM+ nonnaïve CD4 T cells. (F) Representative flow cytometry plots for quantifying AIM+CD8 T cells. The numbers represent the percentage of total nonnaïve CD8 T cells that are AIM+ on days 0 and 42. Below, summary data of AIM+CD8 T-cell frequency before and after vaccination. The values represent the background-subtracted frequency of AIM+ nonnaïve CD8 T cells.
Enhanced vaccine-induced Spike-specific T-cell response in anti-CD19 CAR T cell–treated patients. Healthy individuals (n = 26) and (A) anti-CD19 CAR T cell–treated patients (n = 8) were vaccinated on days 0 and 21 with BNT162b2 mRNA vaccine. Blood samples were taken on days 0, 11, 21, 31, 42, and 90. (B) Levels of neutralizing antibodies. IFN-γ (C) and IL-2 (D) secreted into the plasma after whole blood stimulation with Spike-peptide pool and dimethyl sulfoxide control were quantified. (E) Representative flow cytometry plots for quantifying AIM+CD4 T cells. The numbers represent the percentage of total nonnaïve CD4 T cells that are AIM+ on days 0 and 42. Below, summary data of AIM+CD4 T-cell frequency before and after vaccination. The values represent the background-subtracted frequency of AIM+ nonnaïve CD4 T cells. (F) Representative flow cytometry plots for quantifying AIM+CD8 T cells. The numbers represent the percentage of total nonnaïve CD8 T cells that are AIM+ on days 0 and 42. Below, summary data of AIM+CD8 T-cell frequency before and after vaccination. The values represent the background-subtracted frequency of AIM+ nonnaïve CD8 T cells.
The Spike-specific T-cell response induced by vaccination was longitudinally analyzed at 6 different time points: before vaccination, 10 and 21 days after the first and second dose, and then at 90 days after vaccination. Whole blood was stimulated with a Spike-peptide pool overnight, and secreted interferon γ (IFN-γ) and interleukin-2 (IL-2) were quantified.15
Before vaccination, supernatants of whole blood cultures from CAR T cell–treated patients and healthy controls contained low median IFN-γ (<1.7 pg/mL) and IL-2 (<1.4 pg/mL) levels (Figure 1C-D). Ten days after the first dose, peptide-triggered IFN-γ and IL-2 clearly increased in both groups similarly, in line with the previously demonstrated ability of BNT162b2 to rapidly induce Spike-specific T cells.5,16 In contrast, after the second dose, we observed that the patients with CAR T cells produced quantities of IFN-γ (Figure 1C) and IL-2 (Figure 1D) after Spike-peptide stimulation that were 1 log higher than that detected in healthy controls. We followed 3 of the patients for 180 days after vaccination, and their peptide-induced cytokine release remained high (supplemental Figure 1).
To better define whether the enhanced response to Spike peptides observed after the second dose was derived from Spike-specific CD4 or CD8 T cells, we stimulated peripheral blood mononuclear cells (PBMCs) collected on days 0 and 42 with the Spike-peptide pool for expression of activation markers on gated nonnaïve CD4+ and CD8+ T cells (supplemental Figure 2). We visualized higher frequencies of Spike-specific CD4 T cells in all patients with CAR T cells (median = 4.4%) compared with healthy controls (median = 2.0%; Figure 1E). Similarly, higher median levels of Spike-specific CD8 T cells were also detected in patients with CAR T-cells (healthy median = 0.12%; CAR T cells median = 0.26%), with 3 of 8 showing frequencies that were above that of healthy controls (Figure 1F).
The emergence of highly mutated variants of concern (VOCs) has highlighted that vaccine-induced humoral immunity cannot prevent infection.2,3 Yet, vaccines are still able to provide effective protection from severe COVID-19.4 This can be explained by the ability of cellular immunity to target numerous epitopes along the whole Spike protein and therefore remains largely intact against emerging VOCs.6-9 We thus tested whether vaccine-induced T cells in patients with CAR T cells were also directed against multiple Spike regions.
PBMCs were stimulated with 7 Spike-peptide pools, each covering about 180 amino acids of the Spike protein and analyzed by IFN-γ-ELISpot assay. Figure 2A-C shows the ability of vaccine-induced T cells present in patients with CAR T cells to recognize multiple Spike regions, similar to those in healthy controls. Subsequently, we tested their capacity to respond to SARS-CoV-2 variants B.1.617.2 (Delta) and B.1.1.529 (Omicron). PBMCs were stimulated with 5 peptide pools covering the entire Spike (253 peptides) of the ancestral SARS-CoV-2 and the mutated regions in the Delta (30 peptides) and Omicron (68 peptides) variants, with and without the amino acid substitutions/deletions characteristic for these 2 VOCs. The calculated frequencies of IFN-γ-spot forming cells in response to Spike peptides of the VOCs was compared with the ancestral strain (Figure 2D). Vaccine-induced T-cell immunity was almost completely preserved against Delta in all patients, except in 1 where we detected a reduction of nearly 50% (mean = 9.7% inhibition; Figure 2Ei). Unsurprisingly, the higher mutated Omicron variant did impact the T-cell response more (mean = 20.8% inhibition), yet only in 1 patient did we observe a strong reduction in the T-cell response of nearly 80% (Figure 2Eii). Investigations are underway to identify which T-cell epitopes are affected by Omicron mutations in this individual, who is characterized by the HLA-type A*11:01, A*33:03, B*40:01, B*58:01, C*03:02, C*07:02, DRB1*03:01, and DRB1*11:01.
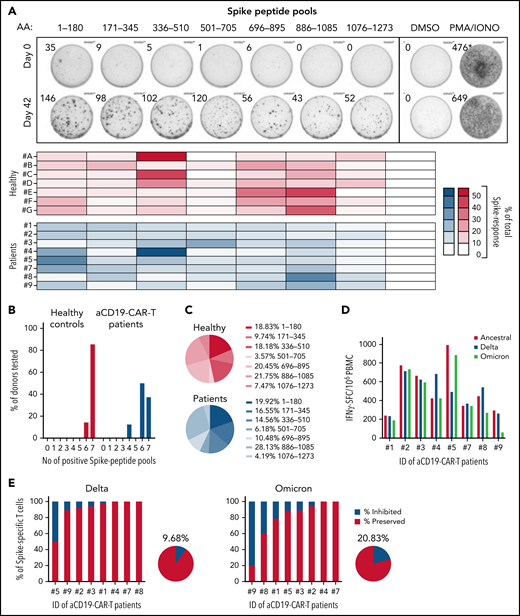
Vaccine-induced Spike-specific T cells are multispecific and preserved against VOCs. (A) PBMCs collected on days 0 and 42 were stimulated with 7 different peptide pools covering distinct regions of the Spike protein and activation was analyzed by IFN-γ-ELISpot assay. Representative ELISpot assay is shown for 1 patient on days 0 and 42. Heatmaps indicate the percentage of the response toward a single peptide pool in proportion to the total Spike-specific response in each of the tested individuals: healthy (red) and CD19 CAR T-cell patients (blue). (B) Bar graphs show the percentage of donors (healthy, n = 7; CD19 CAR T cell, n = 8) reacting to the number of Spike peptide pools tested (total 7 distinct peptide pools). (C) Mean proportion of the response to the 7 distinct Spike peptide pools in healthy (i) and CD19 CAR-T patients (ii). (D) IFN-γ-spot forming cells (SFC) per 1 million PBMC reactive to Spike-peptide pools of the ancestral (red), Delta (blue), and Omicron (green) SARS-CoV-2 variants are shown. (E) Percentage of Spike-specific T cells reactive to the Delta (i) and Omicron (ii) SARS-CoV-2 variants is shown for all 8 patients. The pie charts indicate the mean inhibition.
Vaccine-induced Spike-specific T cells are multispecific and preserved against VOCs. (A) PBMCs collected on days 0 and 42 were stimulated with 7 different peptide pools covering distinct regions of the Spike protein and activation was analyzed by IFN-γ-ELISpot assay. Representative ELISpot assay is shown for 1 patient on days 0 and 42. Heatmaps indicate the percentage of the response toward a single peptide pool in proportion to the total Spike-specific response in each of the tested individuals: healthy (red) and CD19 CAR T-cell patients (blue). (B) Bar graphs show the percentage of donors (healthy, n = 7; CD19 CAR T cell, n = 8) reacting to the number of Spike peptide pools tested (total 7 distinct peptide pools). (C) Mean proportion of the response to the 7 distinct Spike peptide pools in healthy (i) and CD19 CAR-T patients (ii). (D) IFN-γ-spot forming cells (SFC) per 1 million PBMC reactive to Spike-peptide pools of the ancestral (red), Delta (blue), and Omicron (green) SARS-CoV-2 variants are shown. (E) Percentage of Spike-specific T cells reactive to the Delta (i) and Omicron (ii) SARS-CoV-2 variants is shown for all 8 patients. The pie charts indicate the mean inhibition.
Although our study is limited by its small sample size and the use of a single vaccine platform (BNT162b2), our results are reassuring that a stronger induction of Spike-specific T cells after vaccination in patients with anti-CD19 CAR T-cells is seen compared with healthy individuals. Interestingly, this augmented response was only evident after the second dose, similar to observations in vaccinated anti-CD20–treated patients.17 In the absence of vaccine-induced antibody, mRNA-derived Spike antigens might persist longer and were more efficiently presented, resulting in a more robust T-cell response. This hypothesis is consistent with recent data in healthy individuals where low antibody titers were correlated with an enhanced boosting effect from the third vaccine dose.18 This observed enhanced cellular immunity, comprised of both CD4 and CD8 T cells and further characterized by broad specificity against various regions of Spike and its ability to tolerate mutations present in VOCs, might offer some protective efficacy.19 Our results support the clinical utility and importance of COVID-19 vaccination in patients after anti-CD19 CAR T-cell therapy despite B-cell aplasia.
Acknowledgments
The authors thank the patients, families, nurses, and clinical research coordinators who took their time to participate in the study.
This study was supported by the Singapore National Medical Research Council Clinician Scientist Investigator (NMRC/CSA/0053/2008, NMRC/CSA/0053/2013, and NMRC/CSA/MOH/000227), Research Training Fellowship (NMRC/RTF/MOH/000616), Cancer Science Institute, Singapore, and Centre grant (NMRC/CG/NCIS/2010) Goh Foundation, Children’s Cancer Foundation, Singapore Totalisator Board, and VIVA Foundation for Children with Cancer. Moreover, the work was supported by the National Research Foundation (NRF-000230-00 and NRF-CRP25-2020-0003) and the Singapore Ministry of Health’s National Medical Research Council MOH-000019 (MOH-StaR17Nov-0001).
Authorship
Contribution: B.L.Z.O., N.T., A.E.J.Y., A.B., and N.L.B. designed research; N.T., R.d.A., and K.K. performed research; Z.C., M.P., E.C., and J.G.H.L. recruited patients and collected clinical data; N.T. and N.L.B. analyzed data; and B.L.Z.O., N.T., A.B., and N.L.B. wrote the paper.
Conflict-of-interest disclosure: N.L.B. and A.B. reported a patent for a method to monitor SARS-CoV-2–specific T cells in biological samples pending. A.B. reported personal fees from Oxford Immunotech and Qiagen outside the submitted work. The remaining authors declare no competing financial interests.
Correspondence: Nina Le Bert, Emerging Infectious Diseases Program, Duke-NUS Medical School, 8 College Rd, Singapore 169857, Singapore; e-mail: nina@duke-nus.edu.sg.
For original data, please contact nina@duke-nus.edu.sg.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal