

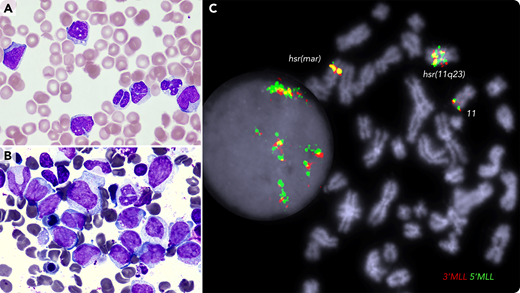
A 72-year-old female with a history of JAK2-mutated essential thrombocythemia presented with easy bruising and hematuria; white blood cell count of 33 × 109/L; monocyte count of 4.95 × 109/L; platelet count of 35 × 109/L; prothrombin time of 17.1 seconds; APTT, 71 seconds; fibrinogen level of 1.74 g/L; D-dimer of 80 000 µg/L FEU; indicative of disseminative intravascular coagulation (DIC). The blood film (panel A; May-Grünwald-Giemsa stain, 100× objective, original magnification ×1000) showed vacuolated blasts, monocytosis, dysplastic neutrophils, red cell fragments, and thrombocytopenia. Bone marrow smear (panel B; May-Grünwald-Giemsa stain, 100× objective, original magnification ×1000) showed large blasts and hypogranulated granulocytes; by flow cytometry, blasts (41%) were positive for CD34, CD33, CD15, CD38, and cMPO. Molecular karyotyping identified a complex genome with chromothrypsis within the 11q23/qter segment, resulting in amplification of many genes, including the MLL gene. Fluorescence in situ hybridization analysis detected MLL amplification and mapped it at the hrs (homogenously stained) regions (panel C; 100× objective, original magnification ×1000; DAPI staining). Next-generation sequencing identified TP53 p.Arg248Gln (VAF 98%), JAK2 p.Val617Phe (VAF 64%), and BCORL1 p.Pro482GlnfsTer16 (VAF 18%) variants. The patient was treated with daily blood products, venetoclax and azacytidine. She died 2 weeks later with refractory leukemia and persistent DIC.
Acute myeloid leukemia with MLL amplification is associated with elderly patients, TP53 mutation, complex karyotype, frequent DIC, an aggressive clinical course, poor response to chemotherapy, and extremely short survival. Clinical trial approach is warranted.
A 72-year-old female with a history of JAK2-mutated essential thrombocythemia presented with easy bruising and hematuria; white blood cell count of 33 × 109/L; monocyte count of 4.95 × 109/L; platelet count of 35 × 109/L; prothrombin time of 17.1 seconds; APTT, 71 seconds; fibrinogen level of 1.74 g/L; D-dimer of 80 000 µg/L FEU; indicative of disseminative intravascular coagulation (DIC). The blood film (panel A; May-Grünwald-Giemsa stain, 100× objective, original magnification ×1000) showed vacuolated blasts, monocytosis, dysplastic neutrophils, red cell fragments, and thrombocytopenia. Bone marrow smear (panel B; May-Grünwald-Giemsa stain, 100× objective, original magnification ×1000) showed large blasts and hypogranulated granulocytes; by flow cytometry, blasts (41%) were positive for CD34, CD33, CD15, CD38, and cMPO. Molecular karyotyping identified a complex genome with chromothrypsis within the 11q23/qter segment, resulting in amplification of many genes, including the MLL gene. Fluorescence in situ hybridization analysis detected MLL amplification and mapped it at the hrs (homogenously stained) regions (panel C; 100× objective, original magnification ×1000; DAPI staining). Next-generation sequencing identified TP53 p.Arg248Gln (VAF 98%), JAK2 p.Val617Phe (VAF 64%), and BCORL1 p.Pro482GlnfsTer16 (VAF 18%) variants. The patient was treated with daily blood products, venetoclax and azacytidine. She died 2 weeks later with refractory leukemia and persistent DIC.
Acute myeloid leukemia with MLL amplification is associated with elderly patients, TP53 mutation, complex karyotype, frequent DIC, an aggressive clinical course, poor response to chemotherapy, and extremely short survival. Clinical trial approach is warranted.
For additional images, visit the ASH Image Bank, a reference and teaching tool that is continually updated with new atlas and case study images. For more information, visit http://imagebank.hematology.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal