

Key Points
We assessed tumor-intrinsic factors driving clinical failures of anti-CD19 CAR T cells through whole-genome sequencing.
Distinct mutational processes, complex structural variants, and deletion of RHOA associated with CAR-19 resistance and early progression.

Abstract
CD19-directed chimeric antigen receptor (CAR-19) T cells are groundbreaking immunotherapies approved for use against large B-cell lymphomas. Although host inflammatory and tumor microenvironmental markers associate with efficacy and resistance, the tumor-intrinsic alterations underlying these phenomena remain undefined. CD19 mutations associate with resistance but are uncommon, and most patients with relapsed disease retain expression of the wild-type receptor, implicating other genomic mechanisms. We therefore leveraged the comprehensive resolution of whole-genome sequencing to assess 51 tumor samples from 49 patients with CAR-19–treated large B-cell lymphoma. We found that the pretreatment presence of complex structural variants, APOBEC mutational signatures, and genomic damage from reactive oxygen species predict CAR-19 resistance. In addition, the recurrent 3p21.31 chromosomal deletion containing the RHOA tumor suppressor was strongly enriched in patients for whom CAR T-cell therapy failed. Pretreatment reduced expression or monoallelic loss of CD19 did not affect responses, suggesting CAR-19 therapy success and resistance are related to multiple mechanisms. Our study showed that tumor-intrinsic genomic alterations are key among the complex interplay of factors that underlie CAR-19 efficacy and resistance for large B-cell lymphomas.
Introduction
Chimeric antigenreceptor-modified T cells targeting CD19 (CAR-19) have recently emerged as an unprecedented therapeutic option for patients with relapsed/refractory (r/r) diffuse large B-cell lymphoma (DLBCL) and related aggressive lymphomas. Although 52% to 82% of patients respond immediately after CAR-19 infusion, only ∼40% achieve complete tumor eradication and prolonged remission,1-5 and underlying mechanisms of treatment failures remain largely unknown. Metabolic tumor volume (MTV) and need for bridging therapy have been associated with poor CAR-19 outcomes, but standard established prognostic factors in DLBCL have little impact.6,7 These findings suggest that, although patients with relapsed or refractory DLBCL share a common histological and clinical phenotype, the underlying biology is distinctly heterogeneous, affecting responses to novel immunotherapies. To decipher this complexity, several groups have focused their investigations on host immune or tumor microenvironment (TME) factors, showing that serum inflammatory markers and T-cell exhaustion in either the TME8 or the CAR-19 product9 are associated with poor outcome.1,7,10 The role of lymphoma genomes in driving these effects has been minimally explored, limited to exome sequencing from formalin-fixed samples collected at various times before CAR-T-cell therapy and suggesting a potential role for mutation of TP53 and other established lymphoma drivers.11,12 Point mutations in CD19 that abrogate CAR binding are demonstrated in individual cases,13 but loss of the CD19 CAR target accounts for only 5% to 30% of relapsed cases,14,15 similar to results for the BCMA CAR target in multiple myeloma.16
We therefore were motivated to dissect the role of genomic drivers involved in thwarting CAR-19 efficacy. We performed whole-genome sequencing (WGS) analysis of 51 large B-cell lymphoma (LBCL) tumors from 49 CAR-19–treated patients. In line with previous evidence, CD19 monoallelic loss and/or low expression by flow cytometry did not associate with resistance, being mostly confined to patients with complete responses and excellent outcome, and 4 of 4 samples analyzed at relapse expressed CD19. Specific genomic findings including apolipoprotein B mRNA-editing enzyme, catalytic (APOBEC) polypeptide mutational activity, deletion 3p21.31 containing RHOA, genomic damage from reactive oxygen species, and complex structural variants (SVs) were significantly associated with CAR-19 treatment failures, suggesting that resistance to this potentially curative immunotherapy is not only driven by immune microenvironment exhaustion but also by distinct cancer cell genomic driver events.
Materials and methods
Patients
Patient characteristics and clinical outcomes for our cohort of 49 patients r/r with LBCL are in Table 1 and supplemental Table 1 (available on the Blood Web site). Germline and tumor samples were collected prospectively under institutional review board protocols8 and in accordance with the Declaration of Helsinki. WGS was performed in patients with adequate samples at the time of analysis without further selection. Durable responders (nonprogressors) were defined as patients who maintained remission after a minimum follow-up of 6 months after CAR-19 infusion. Nondurable responders (progressors) had disease refractory to CAR-19 or lymphoma recurrence after an initial response. To increase the statistical power of specific analyses, we included study 50 patients with newly diagnosed DLBCL from the Pan-Cancer Analysis of Whole Genomes (PCAWG) consortium,17 after removing the sample carrying BRCA mutations.
Patient information
| Characteristic | All patients (N = 49) |
|---|---|
| Median age, y, (range) | 65 (44-79) |
| Sex | |
| Female | 11 (22.4) |
| Male | 38 (77.6) |
| Disease | |
| DLBCL | 40 (81.6) |
| tFL | 8 (16.3) |
| tCLL | 1 (2.0) |
| Stage at apheresis | |
| I/II | 8 (16.3) |
| III/IV | 41 (83.7) |
| IPI at apheresis | |
| 1-2 | 15 (30.6) |
| 3-5 | 34 (69.4) |
| ECOG at apheresis | |
| 0-1 | 38 (77.6) |
| 2-3 | 11 (22.4) |
| Median prior treatment regimens, n (range) | 2 (1-6) |
| Salvage chemotherapies | |
| Platinum compounds | 40 (81.6) |
| Cisplatin | 11 (22.4) |
| Carboplatin | 17 (34.7) |
| Oxaliplatin | 15 (30.6) |
| Melphalan | 11 (22.4) |
| Previous HDT/ASCR | 11 (22.4) |
| Bridging therapy | |
| No | 15 (30.6) |
| Yes | 34 (69.4) |
| CAR-19 product | |
| Axicabtagene ciloleucel | 45 (91.8) |
| Tisagenlecleucel | 2 (4.1) |
| Lisocabtagene maraleucel | 2 (4.1) |
| CRS grade | |
| 0 | 9 (18.4) |
| 1-2 | 36 (73.5) |
| 3-5 | 4 (8.2) |
| ICANS grade | |
| 0 | 16 (32.7) |
| 1-2 | 18 (36.7) |
| 3-4 | 15 (30.6) |
| CAR-19 treatment outcome | |
| Response without progression | 18 (36.7) |
| Response with progression (relapse) | 23 (46.9) |
| Refractory disease | 8 (16.3) |
| Characteristic | All patients (N = 49) |
|---|---|
| Median age, y, (range) | 65 (44-79) |
| Sex | |
| Female | 11 (22.4) |
| Male | 38 (77.6) |
| Disease | |
| DLBCL | 40 (81.6) |
| tFL | 8 (16.3) |
| tCLL | 1 (2.0) |
| Stage at apheresis | |
| I/II | 8 (16.3) |
| III/IV | 41 (83.7) |
| IPI at apheresis | |
| 1-2 | 15 (30.6) |
| 3-5 | 34 (69.4) |
| ECOG at apheresis | |
| 0-1 | 38 (77.6) |
| 2-3 | 11 (22.4) |
| Median prior treatment regimens, n (range) | 2 (1-6) |
| Salvage chemotherapies | |
| Platinum compounds | 40 (81.6) |
| Cisplatin | 11 (22.4) |
| Carboplatin | 17 (34.7) |
| Oxaliplatin | 15 (30.6) |
| Melphalan | 11 (22.4) |
| Previous HDT/ASCR | 11 (22.4) |
| Bridging therapy | |
| No | 15 (30.6) |
| Yes | 34 (69.4) |
| CAR-19 product | |
| Axicabtagene ciloleucel | 45 (91.8) |
| Tisagenlecleucel | 2 (4.1) |
| Lisocabtagene maraleucel | 2 (4.1) |
| CRS grade | |
| 0 | 9 (18.4) |
| 1-2 | 36 (73.5) |
| 3-5 | 4 (8.2) |
| ICANS grade | |
| 0 | 16 (32.7) |
| 1-2 | 18 (36.7) |
| 3-4 | 15 (30.6) |
| CAR-19 treatment outcome | |
| Response without progression | 18 (36.7) |
| Response with progression (relapse) | 23 (46.9) |
| Refractory disease | 8 (16.3) |
Data are expressed as the number of patients (percentage of the study group), unless otherwise indicated.
CRS, cytokine release syndrome; ECOG, Eastern Cooperative Oncology Group; HDT/ASCR, high-dose therapy with autologous stem-cell rescue; ICANS, immune effector cell-associated neurotoxicity syndrome; IPI, International Prognostic Index; tCLL, transformed chronic lymphocytic leukemia; tFL, transformed follicular lymphoma.
Samples
Patient samples were received as frozen peripheral blood mononuclear cells or viably preserved tumor biopsies and then thawed in a 37°C water bath. Samples were spun at room temperature at 3000g for 5 minutes. Cell pellets were washed once with phosphate-buffered saline before being processing for nucleic acid extraction. AllPrep DNA/RNA Mini Kit (cat. no. 80284; Qiagen) was used to extract DNA, and the samples were eluted in water.
WGS
WGS library construction and sequencing were performed at the Center for Genome Technology at the John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, and at the Molecular Genomics Core at Moffitt Cancer Center, both centers using Illumina NextSeq500. First, all DNA samples were evaluated for concentration by fluorometric Qubit assays (Thermo-Fisher) and for integrity by TapeStation (Agilent Technologies). Sequencing libraries were prepared with the TruSeq DNA PCR-free HT sample preparation kit from Illumina. In brief, 1 ug of total genomic DNA was fragmented, using the Covaris LE220 focus acoustic sonicator to a target size of 350 bp. Blunt-end DNA fragments were generated, and size selection was performed with AMPure bead purification (Beckman Coulter). A-base tailing was performed on the 3′ blunt ends, followed by adapter ligation and a bead-based cleanup of the libraries. Final library fragment size was evaluated on the TapeStation (Agilent Technologies) and final molarity quantification was determined by quantitative polymerase chain reaction with adapter-specific primers (Kapa Biosystems) on a Roche Light Cycler. Libraries were normalized to 2.8 nM, and 24 samples were pooled for sequencing on a S4-300 flow cell on the NovaSeq 6000. Paired-end 150-bp reads were generated to yield an average depth of 30× per sample. FASTQ files were generated with the Illumina BCL2FASTQ algorithm and were used for downstream processing.
WGS analysis
Complete details are available in the supplemental Methods.
Raw FASTQ files were uploaded to the Illumina BaseSpace Sequence Hub for downstream processing. Tumor and normal paired samples were aligned against the GRCh38 genome build and somatic single-nucleotide variants (SNVs) and short insertion-deletions variants (indels) were called using the DRAGEN Somatic Pipeline, Version 3.6.3. We performed additional quality filters (supplemental Methods) and applied the dN/dScv method to detect genes under positive selection in r/r cases18 and increased statistical power using samples from PCAWG14,17 compared through 2-tailed Fisher’s exact test and correction for multiple testing using the false discovery rate. Copy number variations (CNVs) were called with the Sequenza Version 3.0.0 algorithm (https://github.com/cran/sequenza), as previously described for DLBCL.19 The genome regions that were significantly modified in our samples were identified using GISTIC (v2.0.23; https://www.genepattern.org). To improve the test’s statistical power, we again compared with DLBCL PCAWG samples (n = 50) using the Gene Pattern Web interface (http://genepattern.broadinstitute.org). To determinate the tumor clonal architecture, we used PyClone-VI (v0.1.0; https://github.com/Roth-Lab/pyclone-vi) and tested for enrichment again with false discovery rate correction. The mutational signature analysis of single-base substitutions (SBSs) was performed as described previously20-22 and as outlined further in supplemental Methods. To detect SVs, deletions, inversion, translocations, and tandem duplications, we used Manta. Complex events, such as chromothripsis, chromoplexy, and templated insertions, were defined after manual inspection, as previously described.23-26
RNA sequencing analysis
The RNA sequencing (RNA-seq) libraries were prepared with the NuGen RNA-Seq Multiplex System (Tecan US) as previously described.8 The libraries were sequenced on the Illumina NextSeq 500 system with a 75-bp end run at 80 to 100 million read pairs per sample. RNA-seq reads were mapped to the reference human genome (GRCh38) using the STAR algorithm (https://github.com/alexdobin/STAR), and the parameters were set to count read numbers per gene while mapping.
Flow cytometry analysis
Flow cytometry analysis was performed on Kaluza Analysis Software v2.1 (Indianapolis, IN). The x- and y-axis parameters were set to Logicle scale. CD19 staining was considered positive when intensity was 103 to 104, dim below 103 but >1 log higher than a double-negative population (lower left quadrant). Expression below this threshold was considered negative for CD19.
Chapuy clustering
Samples were clustered according to the Chapuy clustering system,27 using SV, CNV, and mutation data.
LymphGen clustering
Samples were categorized into subtypes by LymphGen Classifier (https://llmpp.nih.gov/lymphgen/index.php),28 based on SV, CNV, and mutational data.
Statistics
Comparison tests were performed with Fisher’s exact test. Association of categorial variables with progression-free survival (PFS) and overall survival (OS) were performed in a univariable fashion, using Kaplan-Meier curves and a log-rank test. All analyses were performed in R.
Results
Patient cohort
LBCL tumor biopsy specimens (with paired germline samples) of 49 patients treated with CAR-19 were analyzed by WGS (median coverage, 38.7×, range 17.2-76.08; supplemental Table 1). Forty-four samples were collected immediately before CAR-19 therapy. Two patients had samples collected before treatment and at progression after CAR-19 (CAR_39 and CAR_84). Three cases, 1 refractory (CAR_26), 1 with minimal response (CAR_237), and 1 with transient complete remission (CAR_43) had relapse-only biopsy specimens. Although the CAR_26 and CAR_237 primary refractoriness and limited response was most likely linked to preexisting clonal genetic alterations, the initial complete remission followed by progression in CAR_43 may have reflected selection of a distinct subclone whose genomic landscape differed from the pretreatment sample. Therefore, this case was excluded from PFS and OS analyses.
Demographics, disease characteristics, and response to CAR-19 for all patients are summarized in Table 1. All had LBCL: 40 with de novo DLBCL, 8 with transformed follicular lymphoma, and 1 with transformed chronic lymphocytic leukemia. The median age was 65 (range, 44-79); 11 (22.4%) were women. The median number of prior treatments was 2 (range: 1-6), with 40 (81.6%) patients exposed to platinum-containing regimens. Eleven patients (22.4%) had undergone high-dose melphalan-based conditioning and autologous stem-cell rescue. Thirty-four patients (69.4%) had received bridging therapy between apheresis and CAR-19 infusion. After treatment, 4 patients (8.2%) experienced grade 3 or higher cytokine release syndrome, and 15 patients (30.6%) had grade 3 or higher immune effector cell-associated neurotoxicity syndrome. One patient died within a week after infusion as a result of CAR-19 toxicity with unknown disease response (CAR_122). This patient was omitted from PFS but included in OS analyses. Median OS and PFS for the entire cohort were 11.6 and 8 months, respectively (Figure 1A-B; supplemental Figure 1A). Considering only long responders, median follow-up was 17.3 months. Progression-free response was seen in 18 (36.7%) patients (Table 1). Overall, results are comparable to previously reported CAR-19 outcomes.1,2,5,29 Investigating clinical features associated with poor prognosis after CAR-19 (supplemental Table 2), pretreatment MTV, sex, age, and prior lines of treatment associated with higher risk of relapse when considering all patients (P = .048; P = .029; P = .008; and P = .016 respectively). Age and prior lines of treatment confirmed their significance, when considering de novo DLBCL only (P = .004 and P = .02 respectively; Figure 1C-E; supplemental Figure 2A-B; supplemental Table 2). Improved outcome with increased age was also seen by the US Lymphoma CAR T Consortium3 and may reflect selection bias by referring clinicians, but could not be investigated further with these data. In terms of OS, MTV (P = .048) and bridging therapy (P = .012) showed significant association with shorter survival (supplemental Figure 1B).
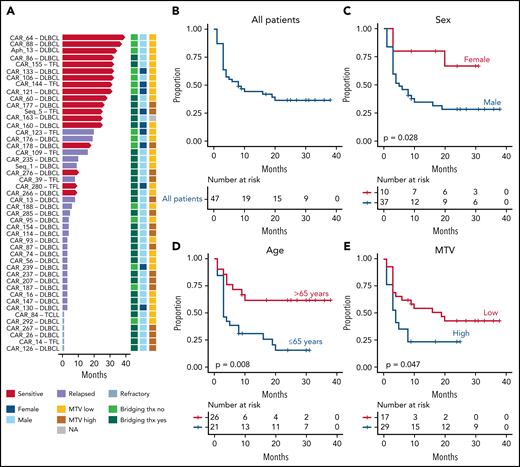
Clinical features associated with early progression in LBCL treated with CAR-19. (A) Follow-up and clinical outcome of 47 of 49 patients with LBCL included in the PFS and OS analysis (excluded cases: relapse-only sample with transitory complete remission and early therapy-related death not evaluable for PFS). Red arrows indicate ongoing responses. (B) Kaplan-Meier curve for PFS for all 47 patients. (C-D) Kaplan-Meier curves for PFS comparing female vs male (C), patients more or less than 65 years old (D), and low vs high MTV (E).
Clinical features associated with early progression in LBCL treated with CAR-19. (A) Follow-up and clinical outcome of 47 of 49 patients with LBCL included in the PFS and OS analysis (excluded cases: relapse-only sample with transitory complete remission and early therapy-related death not evaluable for PFS). Red arrows indicate ongoing responses. (B) Kaplan-Meier curve for PFS for all 47 patients. (C-D) Kaplan-Meier curves for PFS comparing female vs male (C), patients more or less than 65 years old (D), and low vs high MTV (E).
Driver genes associated with CAR-19 outcome in r/r LBCL
We found a median of 12 802 SBSs (range, 2 090-32 857). There was no difference in total mutational burden between progressors and durable responders. To identify driver genes, we leveraged the ratio of nonsynonymous to synonymous mutations using dN/dScv.18 To increase statistical power, we combined our r/r cohort with 50 newly diagnosed DLBCL cases from PCAWG.17 Positive selection was detected for 41 candidate driver genes (q value < 0.1; supplemental Table 3; supplemental Figure 3).18TP53 was the most frequently mutated gene, with 42% of r/r cases containing at least 1 mutation, consistent with previously reports on r/r cases.30 Nevertheless, neither TP53 nor any other individual driver gene predicted poor CAR-19 outcome after correction for multiple hypothesis testing using the false discovery rate (supplemental Table 4).
Prior studies have assessed loss of CD19 as a mechanism of CAR-19 resistance.31 Although individual examples demonstrate lack of response,13 this mechanism of escape seems to explain less than one-third of resistance in DLBCL.14,15 At the genomic level, 3 cases showed a monoallelic copy number loss of CD19, and 2 cases had mutated CD19, 1 of which was subclonal (L174V; 30% cancer cell fraction) and the other, clonal (G210D; 100% cancer cell fraction).32 The latter case also showed a subclonal, focal CD19 deletion (35% cancer cell fraction). Strikingly, 4 of 5 of the patients had durable CAR-19 responses. The case with CD19 L174V is emblematic of the complexity of antitumor activities promoted by CAR-19. An identical mutation was found as clonal at baseline in a patient completely refractory to CAR-19 treatment (Figure 2A, top).13 For our patient, we therefore would have expected a CAR CD19–mediated eradication of CD19 wild-type cells, but not of those harboring CD19 L174V (Figure 2A, middle). Instead, however, CAR-19 infusion induced complete tumor eradication (ie, both CD19 wild-type and CD19 L174V mutated clones) and an ongoing remission of more than 2 years (Figure 2A, bottom). Pretreatment CD19 expression was also tested in 38 cases by flow cytometry, with 8 (10.5%) having absent or reduced protein expression. Although the disease in 6 of 8 patients with reduced or absent CD19 progressed, the correlation between CD19 protein expression and poor outcome was not statically significant (P = .46; supplemental Figure 4). Furthermore, it showed no impact on OS (P = .61; supplemental Figure 4). In addition, 4 of 5 CAR-19 tumors assessed at relapse were CD19+. Finally, transcriptomic investigation of CD19 expression by RNA-seq in 16 patients did not show any impact on either PFS or OS (Figure 2B). Taken together, with 6 of 8 patients lacking CD19 who responded to axicabtagene ciloleucel in the ZUMA-1 registration study2 and recent preclinical studies,33 these findings indicate that antigen-mediated tumor killing is not the only mechanism of tumor eradication.
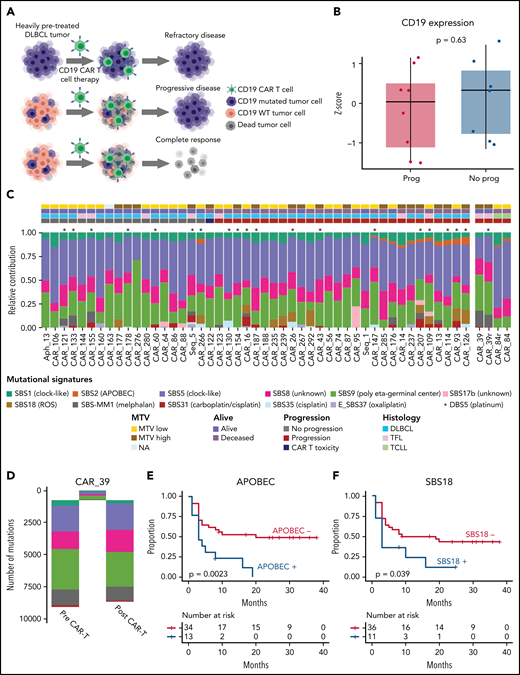
Prognostic impact of SBS mutational signatures in LBCL treated with CAR-19. (A) Summary of all possible impacts of CD19 L174V mutation on tumor evolution after CAR-19. (B) Absence of prognostic impact of CD19 gene expression among 16 patients with available RNA-seq data. (C) The relative contribution of each mutational signature (color) per each sample (x-axis, 51 WGS samples). Asterisks indicate the presence of DBS5 (platinum chemotherapy treatment double-base substitution signature). (D) SBS mutational-signature contributions for each phylogenetic tree cluster (sample CAR_39). SBS mutational signature colors are the same as the legend to panel B. (E-F) The Kaplan-Meier plots of PFS comparing patients with (+) and without (−) APOBEC (E) and SBS18 (F) mutational signatures (n = 47).
Prognostic impact of SBS mutational signatures in LBCL treated with CAR-19. (A) Summary of all possible impacts of CD19 L174V mutation on tumor evolution after CAR-19. (B) Absence of prognostic impact of CD19 gene expression among 16 patients with available RNA-seq data. (C) The relative contribution of each mutational signature (color) per each sample (x-axis, 51 WGS samples). Asterisks indicate the presence of DBS5 (platinum chemotherapy treatment double-base substitution signature). (D) SBS mutational-signature contributions for each phylogenetic tree cluster (sample CAR_39). SBS mutational signature colors are the same as the legend to panel B. (E-F) The Kaplan-Meier plots of PFS comparing patients with (+) and without (−) APOBEC (E) and SBS18 (F) mutational signatures (n = 47).
Mutational signatures associated with CAR-19 outcome in r/r LBCL
To investigate underlying mutational processes (ie, mutational signatures) involved in shaping the repertoire of SBSs, we ran sigProfiler20 and hierarchical Dirichlet.21 To increase resolution and statistical power, we again combined our cohort with WGS from 50 newly diagnosed cases of PCAWG DLBCL.17 Combining these approaches, we identified 12 mutational signatures involved in our cohort of r/r lymphomas (Figure 2C; supplemental Figure 5). Eight of these are currently included in the COSMIC catalog v.2 (https://cancer.sanger.ac.uk/signatures/sbs) and have been reported in newly diagnosed DLBCL: SBS1 (aging), SBS2 (APOBEC), SBS5 (aging), SBS8, SBS9 (poly-η germinal center), SBS13 (APOBEC), SBS17b, and SBS18 (genomic damage from reactive oxygen species).20 The 4 additional extracted signatures are caused by exposure to distinct chemotherapies, and 2 are not yet included in COSMIC (SBS-MM1, melphalan; E_SBS37, oxaliplatin; SBS31, cisplatin/carboplatin; and SBS35, cisplatin signatures; Figure 2C).20,21,34-36 Next, to confirm the presence of each mutational signature and to accurately estimate its contribution, we ran the mmsig fitting algorithm.22 As expected SBS-MM1 mutational signature was identified in 10 of 11 patients who received melphalan as part of high-dose melphalan-based conditioning and autologous stem-cell rescue, in line with prior evidence of the high direct mutagenic impact of melphalan. To accurately define evidence of platinum mutagenic activity, we implemented the double-base substitution analysis (DBS) and detected DBS5 (platinum chemotherapy treatment signature) in 69% (20 of 29) of patients who had evidence of platinum SBS signatures. Interestingly, 7 of 40 (17.5%) previously exposed to platinum did not demonstrate these chemotherapy-related SBS and DBS signatures. It has been shown that distinct chemotherapy agents promote their mutagenic activity by introducing a unique catalog of mutations in each exposed single cell.21,36 Therefore, this single-cell chemotherapy barcode is detectable by bulk WGS, only if a single tumor cell exposed to the chemotherapy expands, taking clonal dominance (ie, in a single-cell expansion model). In contrast, chemotherapy-induced mutational signatures are not detectable if the cancer progression is driven by multiple clones originating from different single cells exposed to chemotherapy and therefore harbors different chemotherapy barcodes (catalog of unique chemotherapy-related mutations). The concept of chemotherapy barcoding can also be used to time events and establish whether the progression was driven by 1 or more single-tumor cells.36 We reconstructed the phylogeny of 2 cases with samples collected before and at relapse after CAR-19 therapy (“Materials and methods”). In 1 patient (CAR_84), the clonal composition did not change over time, and no platinum-related signatures were detected, despite prior exposure, suggesting a complete refractoriness to CAR-19 where the progression is driven by multiple tumor clones. The other case (CAR_39) is an example of branching evolution after CAR-19, with each branch characterized by a unique SBS-MM1 catalog of mutations (Figure 2D). Interestingly this scenario is compatible with progression driven by a single cell previously exposed to melphalan (SBS-MM1) and platinum (SBS31) that took clonal dominance to drive relapse after initial complete remission in response to CAR-T infusion. Overall, these data reveal that, similar to other cancers,36 aggressive lymphomas can increase the mutational burden at relapse related to exposure to mutagenic agents, and progression can be driven by clonal dominance of single surviving cells.
When correlated with CAR-19 response, the presence of APOBEC mutational activity (SBS2 and SBS13) carried significantly worse PFS, with disease in 12 of 13 (92%) patients progressing (P = .0023, all patients; P = .045, only de novo DLBCL; n = 40; Figure 2E; supplemental Figure 2C; supplemental Table 5). APOBEC refers to a family of cytidine deaminases that generates an innate immune response to viruses and that has been shown to be active in many human cancers,20,21 in particular in refractory tumors, in metastasis,37 and in tumors with loss of HLA.38 Specific to lymphoma, APOBEC3 family members have been shown to contribute to lymphomagenesis in primary effusion lymphoma, and its mutagenic activity can be detected in 7.8% of newly diagnosed DLBCL.20,39 Among the additional mutational signatures tested (supplemental Table 5), SBS18 was associated with post–CAR-19 progression in 9 of 11 (81%) patients (P = .0396, all patients; P = .045, only de novo DLBCL; n = 40; Figure 2F; supplemental Figure 2D). This SBS signature reflects genomic damage from oxygen radical stress. Both APOBEC and SBS18 were associated with shortened OS (supplemental Figure 1C-D).
Deletion 3p21.31 (RHOA) and poor CAR-19 responses
We ran GISTIC v2.0 (https://www.genepattern.org)40 to compare the genome-wide CNV distribution between our 49 patients with r/r disease and 50 with newly diagnosed DLBCL in PCAWG17 (“Materials and methods”). We detected 13 arm-level and 10 focal regions of copy number gain and 2 arm-level and 20 focal regions of copy number loss (q value < 0.1; Figure 3A-C). When the prevalence of these significant CNVs was compared between the r/r cases and the newly diagnosed PCAWG cases, the 17p13.1 (TP53) deletion was more frequent in the first group. Monoallelic or biallelic loss of TP53 was highly prevalent (59.2%) but did not carry prognostic impact in patients after CAR-19 (Figure 3B-D). Combining TP53 with the related tumor suppressor CDKN2A, 85.7% of our samples had at least 1 mutated or deleted allele in 1 of the 2 (Figure 3B), reflecting the aggressive nature of the tumors included in our cohort in which the disease had relapsed after multiple courses of intensive chemotherapy. Interestingly, focal-level deletion involving 3p21.31 (RHOA) was the only GISTIC peak strongly predictive of poor outcome after CAR-19 (P = .0013 considering all; P < .0001 considering only de novo DLBCL’ n = 40; Figure 3E; supplemental Figure 2E; supplemental Table 6) with 10 of 11 (91%) patients progressing after CAR-19 infusion. Loss of RHOA also associated with poor OS (P = .023; supplemental Figure 1E). The RHOA protein may affect a wide range of cellular processes in diffuse cell types, and its mechanisms as a DLBCL tumor suppressor remain to be clearly defined. Studies to date show increased motility of malignant and premalignant B lymphocytes in RHOA loss-of-function experiments.41,42 Therefore, dissemination to tissues or niches in the TME that provide sanctuary from CAR-19–initiated immunity is one hypothesis. Given the prevalence of RHOA deletions in newly diagnosed de novo DLBCL27 and now also correlation with poor prognosis to CAR-19, detailed laboratory studies are warranted to explore their role in DLBCL and their impact on CAR-19 responses.
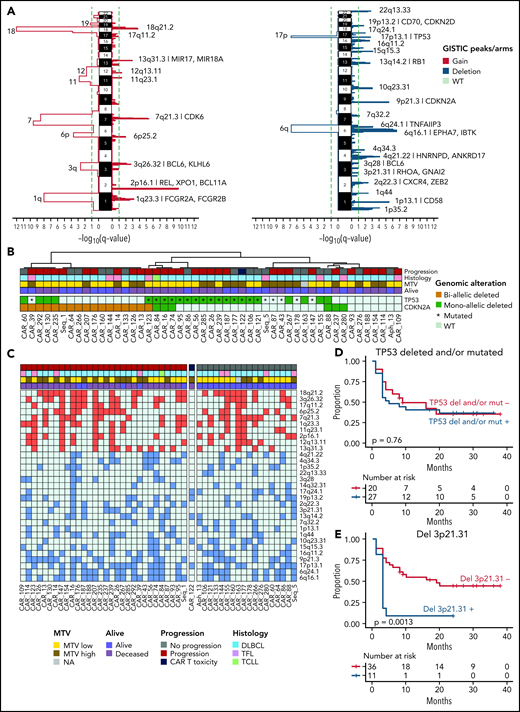
Clinical impact of recurrent copy number anomalies in r/r LBCL. (A) GISTIC significant peaks extracted combining 49 r/r LBCL and samples from 50 PCAWG newly diagnosed cases; amplification (left) and deletion (right). Each plot shows the focal-level and arm-level output; the green dashed line indicates the q-value threshold (q < 0.1). (B) A heat map showing the monoallelic or biallelic alteration of TP53 and CDKN2A in 49 patients with r/r LBCL. (C) A heat map showing the distribution of the 30 focal peaks extracted by GISTIC across 49 patients with r/r LBCL treated with CAR-19. (D-E) Kaplan-Meier plots showing the impact of TP53 loss (D) and deletion 3p21.31 (RHOA) (E) (n = 47).
Clinical impact of recurrent copy number anomalies in r/r LBCL. (A) GISTIC significant peaks extracted combining 49 r/r LBCL and samples from 50 PCAWG newly diagnosed cases; amplification (left) and deletion (right). Each plot shows the focal-level and arm-level output; the green dashed line indicates the q-value threshold (q < 0.1). (B) A heat map showing the monoallelic or biallelic alteration of TP53 and CDKN2A in 49 patients with r/r LBCL. (C) A heat map showing the distribution of the 30 focal peaks extracted by GISTIC across 49 patients with r/r LBCL treated with CAR-19. (D-E) Kaplan-Meier plots showing the impact of TP53 loss (D) and deletion 3p21.31 (RHOA) (E) (n = 47).
Structural variants and poor CAR-19 responses
WGS allows for detailed identification of SVs and complex events. We identified 4011 SVs across the 51 WGS samples (median 60 per r/r sample, range 9-277; Figure 4A). Similar to other hematologic malignancies,23-25,43 we observed evidence of 5 main complex SV event types affecting cases in our cohort: double minutes (n = 6), chromothripsis (n = 23), chromoplexy (n = 18), and templated insertion (n = 11). Chromoplexy is a concatenation of structural variants leading to multiple simultaneous chromosomal losses. Templated insertions represent a concatenation of interchromosomal structural variants leading to a derivative chromosome where multiple focal gains involving oncogenes and regulatory regions are strung together and reinserted in the genome.24,44 Double minutes is a highly amplified region, generally involving distinct oncogenes.43 Chromothripsis represents a catastrophic event in which 1 or >1 chromosome is shattered and aberrantly reassembled generating multiple aneuploidies (Figure 4B).23,44 Templated insertions and chromothripsis were enriched in our cohort compared with PCAWG (P = .015 and P = .034 respectively). Among these complex events, only chromothripsis showed worse PFS (P = .026) after CAR-19 treatment, with 18 of 22 (81.8%) patients experiencing early progression (Figure 4C), but were not associated with OS. Double minutes often involved distinct oncogenes: XPO1 (6 copies in CAR_114, 10 copies in CAR_155), BCL6 (7 copies in CAR_130), CCND3 (5 copies in CAR_26), and MYC (6 copies in CAR_56) (Figure 4D). In all patients with de novo DLBCL with double minutes (4 of 4), the disease rapidly progressed, and the patients died after CAR-19 treatment (P = .017 for PFS Figure 4E; P = .0011 for OS).
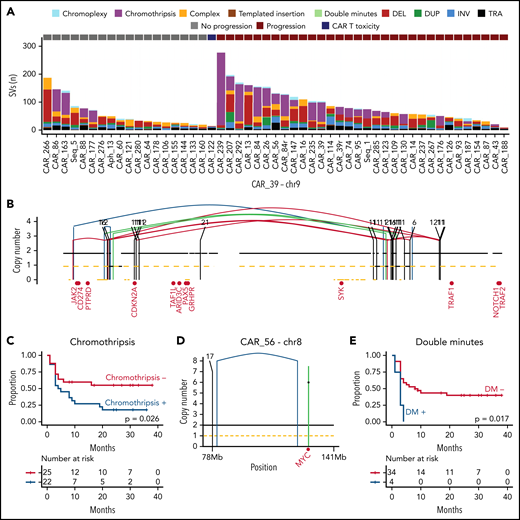
The landscape of structural variants in r/r LBCL and outcome association. (A) Stacked bars show the genome-wide burden of each SV class and complex event per each sample (x-axis, 51 WGS samples from 49 patients). (B) Copy number profile plot integrated with SV information showing an emblematic example of chromothripsis on chromosome 9 responsible of CDKN2A loss (sample CAR_39). The horizontal black line indicates the total copy number; the dashed orange line indicates the minor copy number. The vertical lines represent SV breakpoints, color-coded based on SV class. Red text represents the DLBCL driver genes present on chromosome 9. (C) Kaplan-Meier plot for PFS comparing patients with LBCL, with and without chromothripsis (n = 47) (D-E) Examples of double minutes involving MYC (D) and Kaplan-Meier plot for PFS, comparing patients with DLBCL, with and without double minutes (n = 38).
The landscape of structural variants in r/r LBCL and outcome association. (A) Stacked bars show the genome-wide burden of each SV class and complex event per each sample (x-axis, 51 WGS samples from 49 patients). (B) Copy number profile plot integrated with SV information showing an emblematic example of chromothripsis on chromosome 9 responsible of CDKN2A loss (sample CAR_39). The horizontal black line indicates the total copy number; the dashed orange line indicates the minor copy number. The vertical lines represent SV breakpoints, color-coded based on SV class. Red text represents the DLBCL driver genes present on chromosome 9. (C) Kaplan-Meier plot for PFS comparing patients with LBCL, with and without chromothripsis (n = 47) (D-E) Examples of double minutes involving MYC (D) and Kaplan-Meier plot for PFS, comparing patients with DLBCL, with and without double minutes (n = 38).
Next, we examined the canonical translocations associated with prognosis in previously untreated DLBCL.45 Translocations between immunoglobulin loci and BCL6, BCL2, or MYC did not show any significant prognostic impact, considering either all cases or only de novo DLBCL (supplemental Figure 6A-C; supplemental Table 7). Double hit, defined as cases with a chromosomal rearrangement in MYC together with rearrangement(s) in BCL2 and/or BCL6, did not correlate with outcome (supplemental Figure 6D), nor did double-expression of MYC and BCL2 proteins by routine clinical immunohistochemistry (supplemental Figure 6E). Overall, these observations are in line with recent reports and suggest pathologic features associated with poor outcome in patients with newly diagnosed disease are not prognostic for CAR-19.1,46,47 Leveraging WGS data we combined SV, CNV, and SBS to assign all patients to 1 of the genomic clusters described by Chapuy et al that are predictive of outcome in newly diagnosed DLBCL.27 Most cases fell into cluster 1, characterized by BCL6 and PD1 ligand SVs and NOTCH2 mutations; cluster 2, inactivation of TP53, loss of CDKN2A and RB1 showing perturbed chromosomal stability and cell cycle and recurrent chromosome segment amplification; or cluster 3, BCL2 alterations and mutations in chromatin modifiers, such as KMT2D and CREBBP. Seven patients fell into the more favorable cluster 4, and 1 patient fell into cluster 5, a subgroup with notoriously worse outcome. We also used the publicly available LymphGen classification algorithm,28 and the most of the classified cases fell into the EZB cluster, characterized by epigenetic dysregulation and corresponding to cluster 3 in Chapuy. Neither system showed prognostic significance of CAR-19 treatment (supplemental Figure 6F-G). Overall, these data show that established markers of prognosis in newly diagnosed DLBCL are less predictive of response to CAR-19. On the contrary, the presence of distinct SBS signatures, CNV, and complex SV have emerged as strong and independent predictors of PFS and OS in patients treated with CAR-19 (Figure 5; supplemental Figure 1F). No differences were detected for any of the clinical and genomic features comparing patients with refractory disease (n = 8) with those who had progression after transitory response (n = 23; supplemental Table 8).
![Impact of genomic alterations on clinical outcome. (A) A heat map showing all the genomic alterations associated with progression after CAR-19 cell therapy. (B) Kaplan-Meier plot for PFS, comparing patients with DLBCL with and without any significant genomic drivers (ie, APOBEC mutational activity, double minutes, deletion 3p21.31 [RHOA], SBS18, and chromothripsis; n = 47). (C) Cox multivariate model between the significant clinical and genomic features for PFS.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/5/10.1182_blood.2021015008/6/m_bloodbld2021015008f5.png?Expires=1767701249&Signature=UI2nTGm97AsMWoUekcSC07vzKEb4K179FQAITIPas2eI0CW5-l6AfAn7~psKnRHuS6HmHs6iDhLT-0eZc8yd3gdgu814xGATw7NCweHbEzwtZq2Ak458U7pWwuz8Pqb1wK~-j1EN1Qqp-hGa3A2p-32e1KWYC6VwNOx-axkceRRsBpigcx44EJ4RdPV8Ttdlv9tCdpN8dBUdC9Zjd2hD6GRL3LWS~02yM6TaAH4AfEawDPNwQJhvXxsSuaRMqMNqI7yxtjm5Dwyru6GrDm57nduKkFF1Xd6XkdXRDkS04hYs5ELVcTiUVcb9hEFTBH5SAKL7W5AUddRXsHILFMO2qQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Impact of genomic alterations on clinical outcome. (A) A heat map showing all the genomic alterations associated with progression after CAR-19 cell therapy. (B) Kaplan-Meier plot for PFS, comparing patients with DLBCL with and without any significant genomic drivers (ie, APOBEC mutational activity, double minutes, deletion 3p21.31 [RHOA], SBS18, and chromothripsis; n = 47). (C) Cox multivariate model between the significant clinical and genomic features for PFS.
Impact of genomic alterations on clinical outcome. (A) A heat map showing all the genomic alterations associated with progression after CAR-19 cell therapy. (B) Kaplan-Meier plot for PFS, comparing patients with DLBCL with and without any significant genomic drivers (ie, APOBEC mutational activity, double minutes, deletion 3p21.31 [RHOA], SBS18, and chromothripsis; n = 47). (C) Cox multivariate model between the significant clinical and genomic features for PFS.
Discussion
CAR-T cells,48 next-generation monoclonal antibodies,49 bispecific antibodies,50 and antibody-drug conjugates51,52 are rapidly reshaping clinical management of relapsed and refractory LBCL. Despite unprecedented overall response rates to CAR-19 in heavily pretreated patients, a significant fraction ultimately is failed by these therapies, and this failure almost always associates with poor survival. It is therefore critical to characterize resistance mechanisms to facilitate development of more effective strategies. Most studies published to date have focused on clinical and immune microenvironment features associated with CAR T-cell resistance. At the level of the malignant genome, several reports have shown that loss of the CAR target represents a logical genomic driver of resistance.13,53 However, examining a larger number of patients, for both of the postgerminal center lymphoproliferative disorders where CAR-T cells are approved for use (LBCL and multiple myeloma), such events are uncommon, and the target (CD19 or BCMA) is still expressed in most patients at relapse.14-16 Consistent with these findings, we found that in all post–CAR T samples, CD19 was expressed and that patients with preinfusion CD19 monoallelic/subclonal loss responded. In particular, prolonged remission in a patient with subclonal CD19 L174V mutation supports the concept that CAR-19 antitumor efficacy is also driven by antigen-independent mechanisms. This model has recently been supported by different lines of evidence. For example, multiplex immunostaining of samples from axicabtagene ciloleucel–treated patients showed ≤5% of TME T cells were CAR+ 5 days after infusion, but the CAR− cells were diffusely activated and most likely contributed to both therapeutic efficacy and cytokine release syndrome toxicity.54 Preclinical models specifically demonstrate FAS-mediated killing of antigen-negative tumor cells directly by the CAR-T product.55 Taken as a whole, these data suggest CAR T cells are not just direct cancer cell killers but also provide a gateway into the immunosuppressed TME to allow the host immune system to attack the tumor. Recent studies show the importance of burned-out T cells thwarting immunotherapy in solid tumors56 and that CAR-T cells secrete interferon-γ to activate host T cells in a mouse model of glioblastoma, and these cells preserved their antitumor activity also when infused without CAR T.33 The role of the CAR T cells in invigorating the host immune response in an exhausted TME therefore emerges as a key to maintaining a durable response to CAR T therapy in patients with r/r DLBCL.
Although the role of the immunosuppressed and exhausted microenvironment is undoubtedly critical, we cannot ignore the central role of the cancer cell. In this study, to our knowledge, we have provided the first unbiased genome-wide discovery of cancer cell–intrinsic factors associated with resistance to CAR T-cell therapy. Distinct genomic drivers (ie, RHOA deletions) or genomic complexity indicated by evidence of double minutes, chromothripsis, APOBEC mutational activity, or ROS genomic damage were detected in 26 of 29 (90%) of r/r lymphomas that progressed after CAR T-cell therapy (Figure 5A-B). In contrast to a recent report, our analysis did not reveal TP53 loss as a single-gene driver of CAR-19 outcome.11 The prior study used targeted formalin-fixed, paraffin-embedded sequencing on samples drawn from a mixed population of DLBCL and transformed lymphomas at various times up to more than a year before CAR-19 treatment, which may explain the lower rates of TP53 alteration (37%) in that study compared with our pre-CAR-19 WGS data (59.2%). Overall, the combination of different genomic features and drivers was a much stronger predictor of progression after CAR T-cell treatment than previously reported clinical features, such as MTV (Figure 5C).6 Most of these genomic features do not appear to directly cause a dysregulation of a distinct pathway or gene function, but rather reflect the complexity and ongoing instability of certain genomes. This is similar to other cancers such as myeloma where chromothripsis, APOBEC, and high mutational burden have been linked to poor prognosis after treatment with immunomodulatory agents and proteosome inhibitors.24,57,58 These genomic features, in particular APOBEC, have been linked to distinct microenvironmental exhaustion, genomic instability, immune escape, and inflammatory profiles.38,59 Therefore, we should not see these genomic features as being in opposition to the immune microenvironment, but rather, these data suggest that LBCL CAR T-cell resistance is driven by a complex interplay between immune microenvironment and cancer cells, and this effect must be accounted for in future studies. WGS was necessary for these analyses and is highly impractical in clinical practice and in many research settings as well, requiring fresh-frozen tumors and matched germline DNA. However, we have shown that APOBEC mutational signatures, for example, can be established with exome data.21,22 More importantly, these results can fuel functional studies that establish mechanistically how complex lymphoma genomes promote milieux hostile to CAR T cells and most likely to other emerging immunotherapies.
Acknowledgments
This work was supported by a grant award from the Florida Academic Cancer Center Alliance (J.H.S., M.L.D.); the Sylvester Comprehensive Cancer Center National Cancer Center Institute (NCI) Core Grant P30CA240139 (J.H.S., O.L., F.M.); National Institutes of Health (NIH), NCI Award K23-CA201594 (F.L.L.); and NIH/NCI Ruth L. Kirschstein NSRA Award F30CA265106 (C.A.C.). O.L. has received research funding from NIH, NCI, U.S. Food and Drug Administration (FDA), Multiple Myeloma Research Foundation, International Myeloma Foundation, Leukemia and Lymphoma Society (LLS), Perelman Family Foundation, Rising Tide Foundation. F.M. is supported by ASH. F.L.L. is supported by a Scholar in Clinical Research award from The Leukemia and Lymphoma Society. This research was made possible through the Total Cancer Care Protocol and the Tissue Core and Cell Therapy Facilities at the H. Lee Moffitt Cancer Center and Research Institute, an NCI designated Comprehensive Cancer Center (P30-CA076292) and a donation from the Hyer family in support of CAR-T cell therapy research at Moffitt.
Authorship
Contribution: F.M., F.L.L., J.H.S., and M.L.D. designed and supervised the study, collected and analyzed the data, and wrote the manuscript; M.D.J., B.Z., and C.A.C. collected, analyzed, and interpreted the data and wrote the manuscript; A.J.G., Y.Z., L.C., X.W., and M.H. analyzed the data; O.L., R.F., K.M.R., and M.M. collected the data; and all authors read, revised, and proofed the manuscript.
Conflict-of-interest disclosure: O.L. has received research funding from Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, and Karyopharm; has received honoraria from and served on boards of Adaptive, Amgen, Binding Site, BMS, Celgene, Cellectis, Glenmark, Janssen, Juno, and Pfizer; and serves on independent data monitoring committees (IDMCs) for clinical trials lead by Takeda, Merck, Janssen, and Theradex. M.D.J. reports a consultancy/advisory role for Kite/Gilead, Novartis, Takeda, and BMS, and research funding from Incyte and Kite/Gilead. M.L.D. reports research funding from Celgene, Novartis, and Atara; other financial support from Novartis, Precision Biosciences, Celyad, Bellicum, and GlaxoSmithKline; and stock options from Precision Biosciences, Adaptive, and Anixa. F.L.L. reports a scientific advisory role for Allogene, Amgen, Bluebird Bio, BMS/Celgene, Calibr, Cellular Biomedicine Group, GammaDelta Therapeutics, Iovance, Kite Pharma, Janssen, Legend Biotech, Novartis, Sana, Takeda, Wugen, Umoja; research funding from Kite Pharma (Institutional), Allogene (Institutional), Novartis (Institutional), BlueBird Bio (Institutional), and BMS (Institutional); patents, royalties, and other intellectual property including several patents held by the institution in his name (unlicensed) in the field of cellular immunotherapy; consulting roles for Cowen, EcoR1, Emerging Therapy Solutions, and Gerson Lehrman Group (GLG); and education or editorial activity for Aptitude Health, ASH, BioPharma Communications CARE Education, Clinical Care Options Oncology, Imedex, and Society of Immunotherapy of Cancer. The remaining authors declare no competing financial interests.
Correspondence: Francesco Maura, Myeloma Program, Sylvester Comprehensive Cancer Center, University of Miami, 1120 NW 14th St, Clinical Research Building, Miami, FL 33136; e-mail: fxm557@med.miami.edu; Frederick L. Locke, Division of Clinical Science, Department of Blood and Marrow Transplant and Cellular Immunotherapy, H. Lee Moffitt Cancer Center, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: frederick.locke@moffitt.org; Jonathan H. Schatz, Division of Hematology, Department of Medicine, University of Miami, Batchelor Building, Room 419, Locator M877, 1580 NW 10th Ave, Miami, FL 33136-1000; e-mail: jschatz@med.miami.edu; and Marco L. Davila, Division of Clinical Science, Department of Blood and Marrow Transplant and Cellular Immunotherapy, H. Lee Moffitt Cancer Center, 12902 Magnolia Dr, Tampa, FL 33612; e-mail: marco.davila@moffitt.org.
Submission of raw data to the dbGaP is in process. Pending completion of these uploads, all data is immediately available upon request from the corresponding authors. PCAWG data are available at https://dcc.icgc.org/ and EGAS00001001692 (https://ega-archive.org/studies/EGAS00001001692).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal