Key Points
AML cytogenetic subgroups share a set of 330 altered genes that correlate with myeloid differentiation, leukemic stemness, and relapse.
The unbiased CODEG22 score, including 4 stemness genes and 18 differentiation genes, can help in the risk stratification of AML patients.

Abstract
Advances in transcriptomics have improved our understanding of leukemic development and helped to enhance the stratification of patients. The tendency of transcriptomic studies to combine AML samples, regardless of cytogenetic abnormalities, could lead to bias in differential gene expression analysis because of the differential representation of AML subgroups. Hence, we performed a horizontal meta-analysis that integrated transcriptomic data on AML from multiple studies, to enrich the less frequent cytogenetic subgroups and to uncover common genes involved in the development of AML and response to therapy. A total of 28 Affymetrix microarray data sets containing 3940 AML samples were downloaded from the Gene Expression Omnibus database. After stringent quality control, transcriptomic data on 1534 samples from 11 data sets, covering 10 AML cytogenetically defined subgroups, were retained and merged with the data on 198 healthy bone marrow samples. Differentially expressed genes between each cytogenetic subgroup and normal samples were extracted, enabling the unbiased identification of 330 commonly deregulated genes (CODEGs), which showed enriched profiles of myeloid differentiation, leukemic stem cell status, and relapse. Most of these genes were downregulated, in accordance with DNA hypermethylation. CODEGs were then used to create a prognostic score based on the weighted sum of expression of 22 core genes (CODEG22). The score was validated with microarray data of 5 independent cohorts and by quantitative real time-polymerase chain reaction in a cohort of 142 samples. CODEG22-based stratification of patients, globally and into subpopulations of cytologically healthy and elderly individuals, may complement the European LeukemiaNet classification, for a more accurate prediction of AML outcomes.
Introduction
Acute myeloid leukemia (AML) is a group of genetically heterogeneous hematological malignancies characterized by the accumulation of blasts in the bone marrow (BM), peripheral blood, and other tissues.1 AML is the most common acute leukemia in adults, with a median age of 65 years at diagnosis and an incidence rate ranging between 3 and 5 per 105 patients per year.2,3 Although most patients with AML respond to induction therapy, the global survival rate after 5 years does not exceed 50% for young patients and is <20% for older patients.4 Therapeutic advances may arise from the discovery of genes and pathways that participate in the development and therapeutic resistance of malignant cells, as well as from better patient stratification. According to the World Health Organization and European LeukemiaNet (ELN) recommendations, patients with AML can be stratified based on their underlying genetic defects into 3 risk groups: favorable, intermediate, and adverse. Although advances in genomic technologies have permitted the identification of several somatic mutations (CEBPA, NPM1, FLT3) that have improved stratification of patients,5,6 some patients classified with intermediate and adverse risks do not relapse after therapy and may require adjustment to their treatment. Hence, novel prognostic methods are being proposed to improve risk stratification and guide the decision to treat with intensive chemotherapy or allograft.
Several transcriptomic analyses have been conducted in an attempt to identify the key players in leukemia and to develop prognostic gene expression signatures that can improve treatment.7-12 However, the limited number of patients in individual cohorts has led to the combined analysis of all AML samples, regardless of their cytogenetic abnormalities. Therefore, deregulated genes in rare AML cytogenetic subgroups may have low weight in such global analyses, compared with those from the more frequently occurring subgroups. In addition, extensive studies have been performed to identify prognostic gene expression signatures in leukemic stem cells (LSCs),9,11-14 which are considered key players in resistance to therapy and relapse.15 However, recent studies have demonstrated that relapse in AML does not enrich in cells with LSC capacities and that leukemic-regenerating cells arising after chemotherapy are molecularly distinct from therapy-naive LSCs.16,17 This finding suggests that genes deregulated in AML blasts at diagnosis may harbor valuable prognostic information that is not captured when establishing LSC signatures.
Different cytogenetic abnormalities in AML affect similar biological pathways that are interconnected at the molecular level. Indeed, increased proliferation and inhibition of differentiation are hallmarks of AML, regardless of the underlying genetic defects. In this study, we identified genes and pathways that were consistently deregulated across AML cytogenetic subgroups. For this purpose, we integrated the transcriptomic data of 1534 high-quality AML BM samples, from 11 studies, and 198 healthy control BM samples. In contrast to previous studies that pooled all samples, we separately compared each AML cytogenetic subgroup to the control samples, to determine karyotype-specific differentially expressed genes (DEGs), from which we identified a set of commonly deregulated genes (CODEGs) that was used to create a robust prognostic score. This unbiased score was powerful in the risk stratification of patients with AML in multiple cohorts.
Methods
Data set assembly, quality control, and normalization
Affymetrix data were downloaded as raw CEL files from the Gene Expression Omnibus (GEO) database and were merged into 1 data set (supplemental Methods). The R/Bioconductor18,19 Simpleaffy, and arrayQualityMetrics packages were used to extract quality measurement of the samples,20,21 which were filtered based on exclusion and inclusion criteria.22 High-quality AML samples (n = 1534) were retained by using stringent quality controls and robust multichip average (RMA), normalized with RMAexpress software.
Differential gene expression
In this study, we included in the respective cohorts only AML BM samples that were collected at diagnosis and before any treatments. Pairwise comparisons between each of the 10 AML karyotypes (>10 samples each) and the control normal samples were performed using significance of microarrays23 on samples before and after batch adjustment.24 Cutoffs of log2-fold change >1.5 and value of q < 0.05 were applied for differential gene expression analysis.
Enrichment and protein-protein interaction analyses were performed with Bioconductor’s topGO package,25 the STRING database,26 and Cytoscape software.27 Gene Set Enrichment Analysis (GSEA)28,29 was applied to the normalized data sets GSE76009, GSE65625, and GSE24759. Methylation (HM450) analysis was performed on the AML TCGA (The Cancer Genome Atlas) data set.
Model training and score calculation
Model validation
The score was validated on 5 independent cohorts from 4 microarray data sets, GSE6891,33 GSE10358,34 GSE12417,8 and ALFA-0701,35 using RMA-normalized data.36 The score was also validated on the Beat-AML RNA-sequencing (RNA-seq) data set using transcripts per million–normalized data,37 and by using real-time quantitative polymerase chain reaction (qPCR) on a retrospective cohort of 142 patients from the French Innovative Leukemia Organization (FILO; BB-0033-00073, GOELAMSthèque/FILOthèque, Cochin Hospital, Paris, France). Survival analysis was performed and visualized in the R environment, using the survival38 and survminer39 packages, respectively40 (supplemental Methods).
Results
Characteristics and filtering of downloaded AML samples
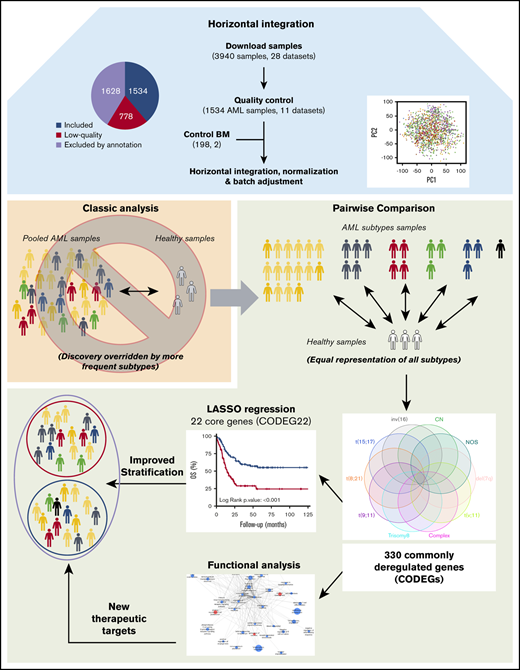
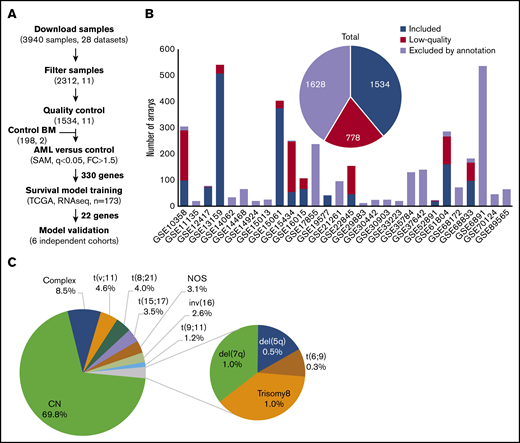
Microarray data for 3940 AML samples (28 data sets), performed on the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array, were downloaded from the GEO database (Figure 1A). This platform was chosen because it is widely available and offers broad genome coverage.41,42 The use of 1 platform is crucial to avoid the variabilities in signal strength that could result from different sensitivities and specificities between probes from different platforms.43 To reduce any factors that may affect gene profiles, only arrays of bulk BM samples, collected from adult patients with AML with known cytogenetics at diagnosis, were considered for further analysis (n = 2312; 11 data sets). We thus excluded samples of unconfirmed origin and those from pediatric AML, peripheral blood, and purified cells (Figure 1B; supplemental Table 1). Raw data from the retained samples were merged and assessed for RNA quality, hybridization quality, and heterogeneity, which resulted in the exclusion of 778 outliers and low-quality samples (Figure 1B; supplemental Figure 1A; supplemental Table 1). The raw data from the remaining 1534 high-quality samples were then merged with a set of 198 unsorted normal BM samples, normalized, and batch adjusted for further analysis (supplemental Figure 1B; supplemental Table 2). The AML samples were representative of 12 different karyotypes, of which the cytogenetically normal (CN) subgroup was the most abundant (69.8%). The t(6;9) and del(5q) samples were excluded because of their low frequency (<10 samples), restricting the analysis to 10 cytogenetic subgroups (Figure 1C; supplemental Table 2). When these stringent filtering steps were followed, the distribution of cytogenetic subgroups was similar to that previously determined by Papaemmanuil et al (supplemental Table 3).5 An extensive multicentric, high-quality data set of cytogenetically diverse AML and normal BM samples was created (GSE147515), offering the opportunity to perform in-depth and powerful analyses.
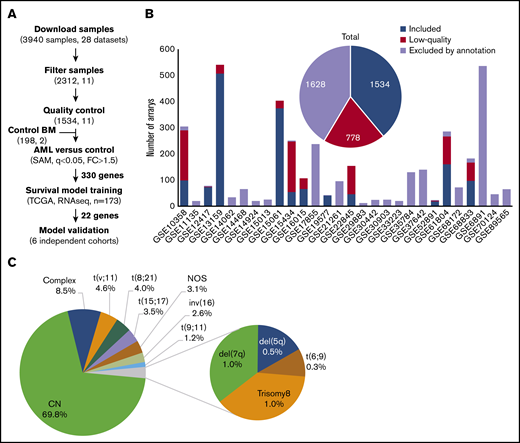
Experimental workflow and sample cytogenetics. (A) Experimental workflow and sample sizes after quality control. (B) Summary of annotation filtering and quality control across data sets. (C) Distribution of the retained samples across cytogenetic abnormalities (n = 1534). NOS, not otherwise specified.
Experimental workflow and sample cytogenetics. (A) Experimental workflow and sample sizes after quality control. (B) Summary of annotation filtering and quality control across data sets. (C) Distribution of the retained samples across cytogenetic abnormalities (n = 1534). NOS, not otherwise specified.
AML subgroups share a common set of deregulated genes and pathways
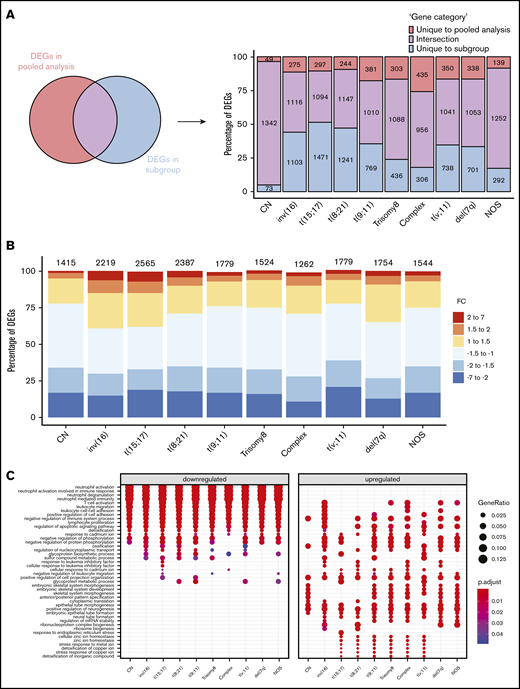
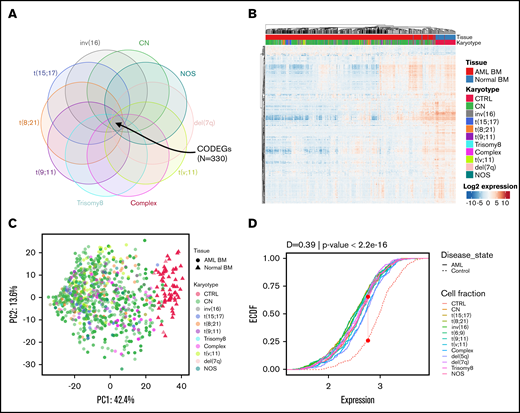
Regardless of their genetic abnormalities, AML subtypes may share common molecular features with therapeutic potentials. To identify common deregulated genes across subgroups, we performed horizontal data integration, which has the capacity to increase both statistical power and material heterogeneity compared with methods used in previous studies.41,44 Indeed, a classic comparison of pooled AML samples, regardless of abnormalities, identified 1391 differentially expressed genes (DEGs; Figure 2A). This approach could be skewed by the overrepresentation of the CN-AML group compared with infrequent cytogenetic subgroups, but analysis of each subgroup separately would prevent such bias. Hence, we performed a pairwise comparison between each of the 10 AML subgroups and normal BM, to identify karyotype-specific DEGs. As expected, most DEGs (96.4%) obtained by the karyotype-specific approach in CN-AML were also identified in the pooled strategy, whereas only 3.6% of the deregulated probes were not captured (Figure 2A). In contrast, between 19% and 57% of the probes deregulated in less frequent subtypes, representing 292 to 1471 probes, were not detected in the pooled approach. More than 60% of DEGs (fold change >1) in each of the cytogenetic subgroups were downregulated in AML, compared with normal BM (Figure 2B). Enrichment analysis of DEGs showed that upregulated and downregulated genes contributed to common biological pathways in all karyotypes (Figure 2C), suggesting that global gene enrichment could be commonly associated with all cytogenetic subgroups. A total of 330 common DEGs (CODEGs) were identified across the 10 AML subgroups (Figure 3A; supplemental Table 4), of which 311 genes were downregulated, whereas 18 genes and 1 noncoding RNA were upregulated in AML subgroups, compared with normal BM. Hierarchical clustering and principal component analysis confirmed that CODEGs were sufficient to differentiate between AML and control samples, but not between AML subgroups (Figure 3B-C). This result indicates that CODEGs are deregulated in association with disease development, independent of cytogenetic abnormalities. The empirical distribution of CODEGs showed consistent left-shifted expression distribution in the AML subgroups, compared with normal BM, indicating a lower expression profile that reflected overrepresented downregulated genes (Figure 3D). Interestingly, the expression profile of CODEGs, particularly of downregulated genes, correlated with their methylation profile and with the mutation status of the methylation regulators (supplemental Results).
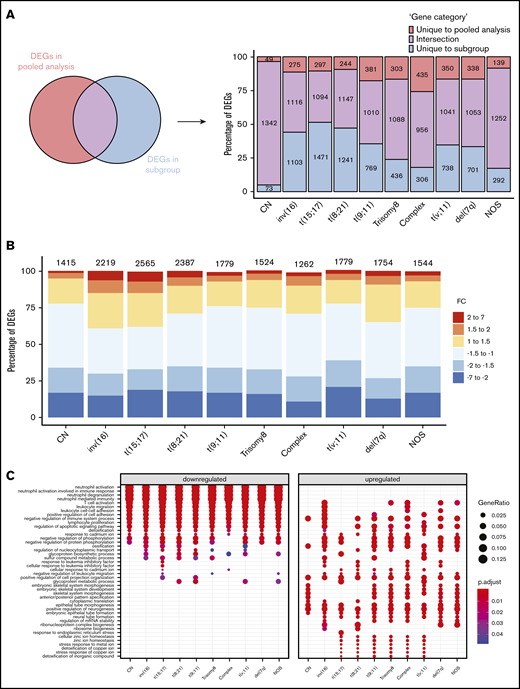
Differential gene expression analysis between AML subgroups and normal BM. (A) Comparison between DEGs in global pooled analysis and group-wise analysis. (B) Fold-change distribution in each cytogenetic subgroup, compared with normal BM samples. (C) Gene Ontology enrichment analysis showing the top pathways associated with upregulated and downregulated genes in each karyotype.
Differential gene expression analysis between AML subgroups and normal BM. (A) Comparison between DEGs in global pooled analysis and group-wise analysis. (B) Fold-change distribution in each cytogenetic subgroup, compared with normal BM samples. (C) Gene Ontology enrichment analysis showing the top pathways associated with upregulated and downregulated genes in each karyotype.
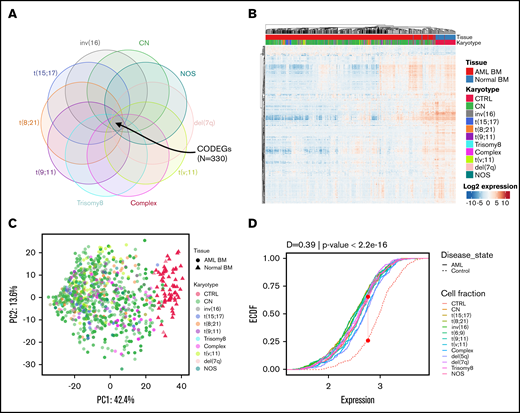
Identification of 330 CODEGs across cytogenetic subgroups. (A) Schematic Venn diagram highlighting the CODEGs. (B) Heat map showing the expression levels of CODEGs in AML and normal BM samples. Centered gene expression is represented in a blue (low expression) to red (high expression) color gradient. Hierarchical clustering of samples was performed using Euclidean distance as a dissimilarity measure and average linkage, and that of genes was performed with Pearson correlation and average linkage. (C) Principal component analysis of samples based on the expression profile of the 330 commonly deregulated genes. Colors represent karyotypes, with red for control samples. (D) Empirical cumulative distribution (ECDF) for the expression of the 330 commonly deregulated genes across AML subgroups. Red points indicate the maximum distance between expression in the control and AML samples. The Kolmogorov-Smirnov goodness-of-fit test was used to test the similarity of distribution between control and AML samples.
Identification of 330 CODEGs across cytogenetic subgroups. (A) Schematic Venn diagram highlighting the CODEGs. (B) Heat map showing the expression levels of CODEGs in AML and normal BM samples. Centered gene expression is represented in a blue (low expression) to red (high expression) color gradient. Hierarchical clustering of samples was performed using Euclidean distance as a dissimilarity measure and average linkage, and that of genes was performed with Pearson correlation and average linkage. (C) Principal component analysis of samples based on the expression profile of the 330 commonly deregulated genes. Colors represent karyotypes, with red for control samples. (D) Empirical cumulative distribution (ECDF) for the expression of the 330 commonly deregulated genes across AML subgroups. Red points indicate the maximum distance between expression in the control and AML samples. The Kolmogorov-Smirnov goodness-of-fit test was used to test the similarity of distribution between control and AML samples.
CODEGs correlate with myeloid differentiation, LSC status, and relapse
Protein-protein interaction analysis showed that 75% of CODEGs were highly interconnected with many downregulated hub genes (supplemental Figure 2A). To identify altered biological processes across AML subgroups, we performed functional enrichment analysis separately on upregulated and downregulated CODEGs. The data showed that upregulated CODEGs were involved in positive regulation of cell proliferation and embryo development, whereas downregulated genes were enriched in pathways related to hematopoietic differentiation and immune responses (supplemental Figure 2B; supplemental Table 5). Therefore, we investigated changes in the expression of CODEGs during normal myeloid differentiation by applying GSEA to the GSE24759 data set.45 Interestingly, upregulated CODEGs were consistently enriched in stem and progenitor cells, whereas downregulated CODEGs were enriched in more mature populations (Figure 4B; supplemental Table 6). Using FAB (French-American-British) information, available for 268 AML samples, we analyzed the average expression profile of upregulated and downregulated CODEGs throughout AML maturation. The average expression of upregulated genes was shown to decrease gradually in AML throughout maturation (supplemental Figure 3). This is consistent with the fact that upregulated genes were increasingly depleted throughout normal myeloid differentiation. In contrast, the variability between FAB subtypes was less pronounced for downregulated genes, where M6 showed higher expression than the M0 to M5 subtypes (supplemental Figure 3). Furthermore, we evaluated the correlation of CODEGs with LSC status in the GSE76009 data set.12 The results showed that upregulated CODEGs were enriched in leukemic stem cells with engraftment potential (LSC+), whereas downregulated CODEGs were enriched in cells with no engraftment potential (LSC−; Figure 4C). Moreover, we compared upregulated and downregulated genes in the CODEG signature to functionally defined hematopoietic stem cells (HSCs) and LSC signatures.9,12 Collectively, the Venn diagrams revealed that upregulated CODEGs contain 4 LSC-related genes (FLT3, SPINK2, TGIF2, and CDK6), of which 2 genes (FLT3 and SPINK2) are also HSC related (supplemental Figure 4). On the other hand, only 16 of 311 downregulated CODEGs were shown to be downregulated in LSCs, including GZMB, which is part of our score. Together, these data indicate that upregulated CODEGs are mostly AML-related genes, whereas downregulated CODEGs, which are enriched during myeloid differentiation, do not overlap with HSC-related genes. To determine whether upregulated genes could be markers of chemoresistance and aggressiveness, we performed GSEA on the GSE66525 data set46 that contained paired diagnosis and relapse AML samples. Remarkably, upregulated CODEGs were enriched in AML samples at relapse, whereas downregulated genes were enriched in samples at diagnosis (Figure 4D). Moreover, we identified the core genes that were positively enriched in relapse samples from the GSE6652546 and GSE8353347 data sets and compared them with the CODEG22 genes. Taking all analyses together, among the 18 upregulated CODEGs identified (supplemental Figures 5 and 6), 7 genes (PLEKHA5, DNM1, MLLT11, CDK6, RABEP2, DNMT3A, and TGIF2) were enriched at relapse in both the GSE66525 (supplemental Figure 5) and GSE83533 (supplemental Figure 6) data sets. Among those, only PLEKHA5 was present within the CODEG22 subset. This gene correlated with good prognosis in the training data set and had a negative coefficient in the score. In contrast, SPINK2 did not increase at relapse, and yet, it had positive coefficients in CODEG22 score and correlated with poor prognosis across multiple data sets.
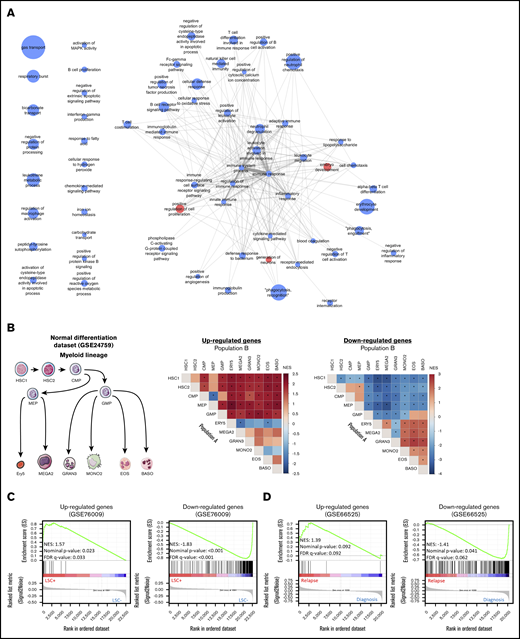
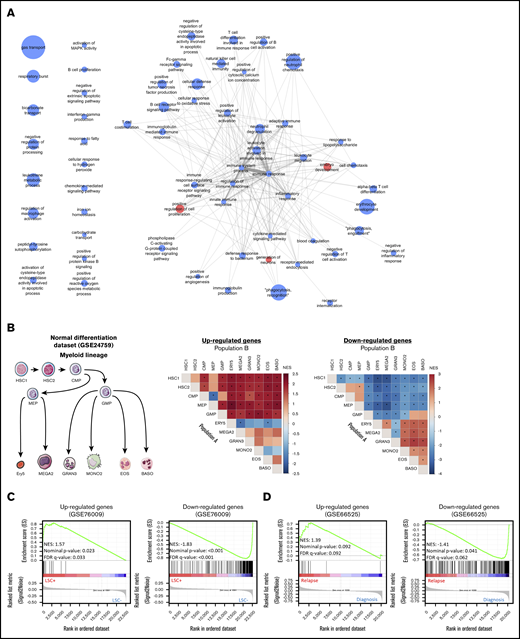
Enrichment analyses of the CODEGs in AML. (A) A network of Gene Ontology biological processes that are enriched in upregulated (red nodes) and downregulated (blue nodes) genes. Node size is proportional to fold enrichment, and edge width and transparency are proportional to the number of shared CODEGs. (B) GSEA of CODEGs throughout normal myeloid differentiation (GSE24759 data set). A differentiation hierarchy representing analyzed hematopoietic populations (left); heat maps summarizing GSEA results on upregulated and downregulated CODEGs, respectively. For each square in the heat maps, a ranked list of DEGs between denoted populations A and B was generated, the enrichment of upregulated (middle) and downregulated (right) CODEG genes was examined against the ranked list, and an enrichment score was generated. Heat map colors correspond to the normalized enrichment score in population A vs B. *Significant enrichments with nominal P < .05 and false discovery rate <0.05. HSC1, CD133+ CD34dim HSCs; HSC2, CD38− CD34+ HSCs; CMP, common myeloid progenitor; GMP, granulocyte/monocyte progenitor; MEP, megakaryocyte/erythroid progenitor; GRAN3, granulocyte (neutrophil); EOS, eosinophil; BASO, basophil; MONO2, monocyte, Ery5, CD34− CD71− GlyA+ erythroid; and MEGA2, megakaryocyte. (C) GSEA of upregulated and downregulated genes in leukemic stem cell positive (LSC+) vs negative (LSC−) populations (GSE76009 data set). (D) GSEA of upregulated and downregulated genes in AML samples at diagnosis vs relapse (GSE66525).
Enrichment analyses of the CODEGs in AML. (A) A network of Gene Ontology biological processes that are enriched in upregulated (red nodes) and downregulated (blue nodes) genes. Node size is proportional to fold enrichment, and edge width and transparency are proportional to the number of shared CODEGs. (B) GSEA of CODEGs throughout normal myeloid differentiation (GSE24759 data set). A differentiation hierarchy representing analyzed hematopoietic populations (left); heat maps summarizing GSEA results on upregulated and downregulated CODEGs, respectively. For each square in the heat maps, a ranked list of DEGs between denoted populations A and B was generated, the enrichment of upregulated (middle) and downregulated (right) CODEG genes was examined against the ranked list, and an enrichment score was generated. Heat map colors correspond to the normalized enrichment score in population A vs B. *Significant enrichments with nominal P < .05 and false discovery rate <0.05. HSC1, CD133+ CD34dim HSCs; HSC2, CD38− CD34+ HSCs; CMP, common myeloid progenitor; GMP, granulocyte/monocyte progenitor; MEP, megakaryocyte/erythroid progenitor; GRAN3, granulocyte (neutrophil); EOS, eosinophil; BASO, basophil; MONO2, monocyte, Ery5, CD34− CD71− GlyA+ erythroid; and MEGA2, megakaryocyte. (C) GSEA of upregulated and downregulated genes in leukemic stem cell positive (LSC+) vs negative (LSC−) populations (GSE76009 data set). (D) GSEA of upregulated and downregulated genes in AML samples at diagnosis vs relapse (GSE66525).
Regularized linear regression applied to CODEGs generates a prognostic score in AML
Because CODEGs correlated with LSC status and response to therapy, we used them to establish a prognostic signature of core genes that correlated with clinical outcomes. The TCGA AML data set was chosen to train the model because, unlike other available data sets, it was obtained by RNA-seq,48 which offers a better representation of gene expression independent of microarray platform. Hence, the LASSO algorithm was applied, using CODEGs as initial predictors. This method generated a simple prognostic AML signature calculated for each patient based on the combined weighted expression of 22 genes, CODEG22 (supplemental Table 7).
Importantly, in the training cohort, patients with a high CODEG22 score showed shorter overall survival (OS) and event-free survival (EFS), than did patients with low score (Table 1: OS time: 8 vs 56.3 months; EFS time: 5.8 vs 20.8 months). The CODEG22 score was not associated with sex, white blood cell count, percentage of blasts, NPM1 mutation or the internal tandem duplication of the FLT3 gene (FLT3-ITD). However, patients with high CODEG22 score had higher median age, lower incidences of favorable cytogenetics, and higher incidences of poor cytogenetics (Table 1).
Recently, ELN recommended the inclusion of ASXL1, TP53, and RUNX1 molecular mutations in prognostications for patients with AML.6 Hence, we validated the CODEG22 score using the new ELN guidelines on the TCGA data set, which has whole-exome sequencing data. Our score remained prognostic after accounting for these mutations, and it improved the prognostic power of the multivariate model containing ELN molecular mutations (FLT3-ITD, NPM1, biallelic-CEBPA, ASXL1, TP53, and RUNX1), cytogenetic abnormalities, age, and WBC. We also verified the correlation of CODEG22 with a wide spectrum of recurrent AML mutations covering many functional groups (supplemental Table 8). Fisher’s exact test revealed that CODEG22 did not correlate with most of these mutations, except for TP53 and WT1 mutations, which were enriched in the high- and low-score groups, respectively. Nonetheless, the score remained prognostic in a multivariate analysis after adjustment for these mutations and other risk factors (supplemental Tables 9 and 10).
The prognostic power of CODEG22 was independently validated on 2 well-annotated AML microarray data sets, which include various cytogenetic subgroups (supplemental Results). Indeed, a high CODEG22 score correlated with poor OS and EFS (Figure 5A-B). The model retained its prognostic significance in the cytogenetically abnormal subset (Figure 5C-D), and it remained prognostic after adjustment for age and cytogenetic abnormalities (supplemental Results). Likewise, the score was also validated on the Beat-AML RNA-seq data set (supplemental Results).
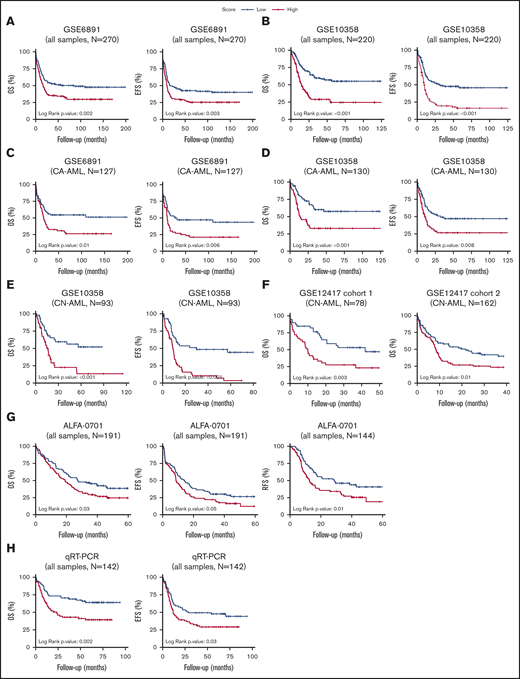
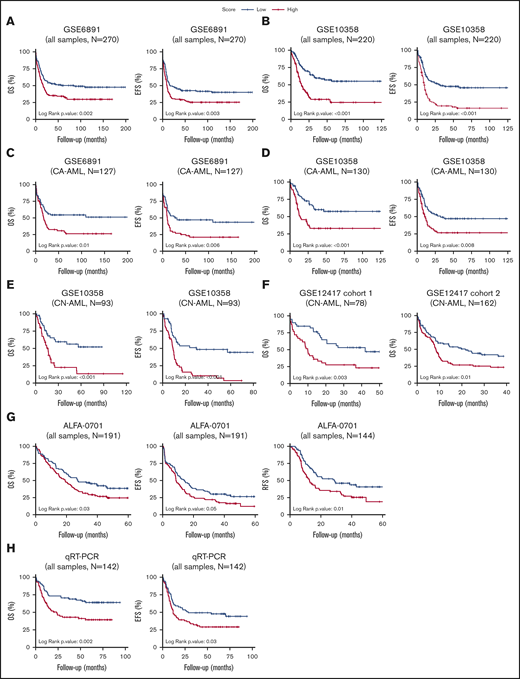
Patients with AML stratified based on high and low CODEG22 score. (A-B) OS and EFS of patients including all cytogenetic abnormalities from the GSE6891 (A) and the GSE10358 (B) data sets. (C-D) OS and EFS of patients with CA-AML from the GSE6891 (C) and GSE10358 (D) data sets. (E) OS and EFS of patients with CN-AML from the GSE10358 data set. (F) OS of all patients from CN-AML cohorts 1 and 2 available in the GSE12417 data set. (G) OS, EFS, and RFS of all patients from the ALFA-0701 cohort. (H) OS and EFS of patients with AML from the retrospective real-time qPCR (qRT-PCR) FILO cohort (n = 142). CODEG22 scores above and below the median in each cohort are labeled high (in red) and low score (in blue), respectively. A log-rank test was used to compare the survival curves of the high and low score subsets.
Patients with AML stratified based on high and low CODEG22 score. (A-B) OS and EFS of patients including all cytogenetic abnormalities from the GSE6891 (A) and the GSE10358 (B) data sets. (C-D) OS and EFS of patients with CA-AML from the GSE6891 (C) and GSE10358 (D) data sets. (E) OS and EFS of patients with CN-AML from the GSE10358 data set. (F) OS of all patients from CN-AML cohorts 1 and 2 available in the GSE12417 data set. (G) OS, EFS, and RFS of all patients from the ALFA-0701 cohort. (H) OS and EFS of patients with AML from the retrospective real-time qPCR (qRT-PCR) FILO cohort (n = 142). CODEG22 scores above and below the median in each cohort are labeled high (in red) and low score (in blue), respectively. A log-rank test was used to compare the survival curves of the high and low score subsets.
CODEG22 stratifies patients with AML with CN karyotype
As mentioned, normal cytogenetics represent >50% of de novo AML. The mutational status of NPM1, CEBPA, and FLT3 genes has recently helped in stratification of patients in this AML subgroup.6 However, a high proportion of patients with CN-AML do not carry these mutations and are considered to have intermediate risk. The CODEG22 score efficiently stratified patients with CN-AML from the GSE10358 data set (Figure 5E; OS and EFS, P < .001) and remained efficient after adjustment for age, NPM1 mutation, and FLT3-ITD status (supplemental Table 11). Moreover, inclusion of CODEG22 score in the model containing age, NPM1 mutation, and FLT3-ITD status (multivariate model 1) strongly improved its predictive power (supplemental Table 11; OS likelihood ratio test [LRT] P = 4.2 × 10−5; EFS LRT P = 2 × 10−5). The CODEG22 score was also tested on 2 CN cohorts from the GSE12417 data set,8 annotated only for age and OS. A high CODEG22 score was significantly associated with poor OS in both cohorts (Figure 5F; cohort 1: OS P = .003; cohort 2: OS P = .01), with shorter median OS time (supplemental Table 12) and with poorer survival probability (supplemental Table 13), compared with the low-score group, which remained significant after adjustment for age (supplemental Table 13; cohort 1: OS hazard ratio [HR] = 2.35, P = .005; OS HR = 1.52, P = .038). Altogether, these results indicate that the CODEG22 score is prognostic of patients with CN-AML, independent of age and NPM1/FLT3-ITD status.
CODEG22 stratifies elderly patients with AML
Refinement of the prognosis is particularly challenging for elderly patients with AML who poorly respond to chemotherapy. The robustness of the CODEG22 score was examined in the ALFA-0701 data set that includes patients with AML, mainly >50 years old.35 In this phase 3 clinical trial, patients were treated with classic chemotherapy either alone or in combination with Gemtuzumab ozogamicin. Results showed that the CODEG22 score was independent of sex, age, WBC, karyotype, cytogenetic risk, cytogenetic abnormalities, treatment arm, NPM1 mutation, or FLT3-ITD status (supplemental Table 14). It is noteworthy that 70.8% of patients with a high score relapsed after therapy, compared with 52.8% of patients with a low score. Indeed, the high-score group had more adverse relapse-free survival (RFS), OS, and EFS, than did the low-score group (Figure 5G), with shorter median survival times (supplemental Table 14; RFS: 12.5 vs 28.7 months, P = .01; OS: 19.2 vs 27.3 months, P = .029; and EFS: 8.6 vs 14.1, P = .047) and poorer survival probability (supplemental Table 15; RFS HR = 1.74, P = .012; OS HR = 1.53, P = .024; and EFS HR = 1.42, P = .041). In addition, the CODEG22 score remained prognostic after adjustment for age, treatment, and cytogenetic risk (supplemental Table 15; RFS HR = 1.81, P = .008; OS HR = 1.52, P = .028; and EFS HR = 1.53, P = .016) and enhanced the predictive capacity of the multivariate RFS model (supplemental Table 15; LRT result improved from .216 to .024). These results show that CODEG22 score can be a valuable marker to aid in stratifying elderly patients with AML.
CODEG22 score stratifies intermediate and adverse risk group patients
The cytogenetically diverse GSE61885 and GSE10358 data sets were split into favorable, intermediate, and adverse risk groups, and the prognostic power of CODEG22 score was examined within these populations. Results showed that our score could identify good responders to therapy within the intermediate and adverse groups from both data sets (supplemental Figure 7). However, the score could not identify poor responders within the favorable subtype (data not shown).
CODEG22 score outperforms the LSC17 score in survival prediction
So far, the most efficient prognostic score is based on upregulated genes in the LSC+ compared with the LSC− population.12 The LSC17 score, based on 17 genes, was trained on the microarray GSE6891 data set, whereas the CODEG22 score was trained on the TCGA RNA-seq data set. Therefore, the GSE10358, an independent microarray data set with a broad range of annotations (supplemental Table 16), was used to compare the 2 scores. Global univariate analysis of EFS showed equal significance of the 2 scores (Wald P < .001), with higher HR for CODEG22 compared with LSC17 (supplemental Table 17; HR, 2.31 vs 1.95). The 2 scores remained significant in a multivariate OS model containing both of them, in addition to age and cytogenetic abnormalities. However, the CODEG22 score outperformed the LSC17 score in a multivariate EFS model containing the 2 scores, age, and cytogenetic abnormalities (supplemental Table 17; CODEG22: EFS HR, 1.63, P = .016; LSC17: EFS HR, 1.45, P = .064). Moreover, univariate analysis of the CN-AML subgroup showed that the LSC17 score was less powerful than the CODEG22 score in predicting both OS (HR, 2.15 vs 2.63) and EFS (HR, 1.98 vs 2.85; supplemental Table 18). Only the CODEG22 score remained significant in multivariate models containing the 2 scores, age, and NPM1/FLT3-ITD status (supplemental Table 18; OS HR, 1.94, P = .037; EFS HR, 2.23, P = .007). Overall, these results show that the CODEG22 score is independent of LSC content and could be a stronger prognostic predictor of AML survival than LSC17.
CODEG22 score is validated by real-time qPCR in a retrospective cohort
Given the global robustness of the CODEG22 score on public transcriptomics data sets, we sought to validate it retrospectively by using real-time qPCR on an AML cohort from the FILO. A total of 142 BM samples from patients with AML belonging to different risk groups were analyzed (supplemental Table 19). Although the model correlated with risk groups and age, it was independent of sex and percentage of blasts (supplemental Table 20). It was prognostic on both OS and EFS (Figure 5H; OS log-rank P = .002; EFS log-rank P = .03), with shorter OS and EFS times (supplemental Table 20; OS median time of 23.5 months vs not reached; P = .002; EFS median time of 11.7 vs 28.3 moths, P = .032) and poorer survival probability (supplemental Table 21; OS HR, 2.13, P = .003; EFS HR, 1.58, P = .033) for the high CODEG22 score compared with the low score. These results show that the score is unbiased, independent of microarray platforms and could be implemented in clinical practice.
Discussion
Relapse remains a major limitation in the treatment of AML, particularly in elderly patients. Nevertheless, discovery of driver genes and improved patient stratification, based on recent advances in transcriptomics, have refined the treatment in many cases. Although next-generation sequencing progressively supplants the use of microarray platforms, the large amount of microarray data sets accumulated during the past 2 decades remains an extensive mine of unexploited information for identifying genes and pathways sustaining aggressiveness and chemoresistance of malignant cells. The true originality of our meta-analysis lies in the use of a horizontal integration strategy to create a large data set (GSE147515) that combines AML samples from multiple studies.
Our method allowed for investigation of common features of differential gene expression across multiple cytogenetic AML subgroups, compared independently and separately to normal BM samples. The investigation required a large number of AML samples that would be accessible only through horizontal integration of data from multiple studies. Although RNA-seq is the current method of choice for high-throughput transcriptomic data analysis, the number of public RNA-seq data sets for AML remains small. On the contrary, horizontal data integration is feasible with Affymetrix microarrays. In this study, we used the Affymetrix GeneChip Human Genome U133 Plus 2.0 Array for this purpose because of its wide availability and well-known genomic coverage (>1500 well-annotated and high-quality samples). In contrast, our prognostic model was developed using RNA-seq data and was validated using both microarray and real-time qPCR, proving that the method is platform independent.
Although data for ∼4000 AML samples were publicly available in the GEO database, ∼20% of the samples had to be excluded for lack of annotation. Moreover, almost all CN-AML samples lacked annotation for mutations in the FLT3, NPM1, and CEBPA genes, which would have been otherwise incorporated. In fact, most data sets lack sufficient clinical annotation for reanalysis, raising an alert on the limited availability of clinical data associated with public data sets.49 Nevertheless, thanks to the high number of data sets, we were able to assemble a large cohort of 1732 AML and normal BM samples. Usually, classic comparison of pooled AML samples results in the overrepresentation of CN samples, whereas genes from many infrequent cytogenetic subgroups remain underrepresented. To overcome this problem, we performed pairwise comparisons between each of the 10 AML subgroups with normal BM and identified karyotype-specific DEGs, among which the expression of 330 genes were commonly altered in all subgroups.
Remarkably, most of the CODEGs were downregulated, suggesting a possible epigenetic regulation. Increased DNA methylation has been associated with AML progression.50,51 In agreement, we observed high DNA methylation of most downregulated CODEGs. This result could be explained by the decline of diverse mature cells and blockage of differentiation of blasts. Unfortunately, the absence of DNA methylation data for normal BM prevents a clear validation of our assumption. However, hypermethylation of CpG islands during AML progression depends on DNMT3A,52 which was present among the upregulated CODEGs. According to the literature, inactivating mutations in DNMTs correlated with decreased methylation, but only for 10% of CODEGs. These data further support the concept of increased hypermethylation in AML, which reinforce the possibility of using DNA hypomethylating agents for karyotype-independent treatment of AML.53
Despite the tendency toward gene downregulation, 19 CODEGs were upregulated and thus may be therapeutic targets (supplemental Results). Among these, DNMT3A and FLT3 are frequently mutated and are hallmarks of high-risk AML.5 FLT3, SPINK2, and CDK6 have been found to be upregulated in LSCs.12 MLLT11 and ANKRD28 have been identified in rare chromosomal translocations, whereas CDK6 and SOX4 have been experimentally linked to AML (supplemental Data). More interestingly, other genes were either listed among upregulated genes in transcriptomic analyses (ATP6V0A2, PDGFC, RABEP2, SINHCAF, and TGIF2), but were never investigated, or never described in AML (DNM1, MIB1, NRXN2, PLEKHA5, ZBTB8A, and ZBTB10) (supplemental Data). The latter merit further functional investigation, for instance, by gene silencing in stem cells, as described for the genes identified in the LSC signature.54 This recommendation is supported by the fact that upregulated CODEGs were found upregulated in LSCs compared with bulk AML, whereas downregulated genes showed the opposite. In hematopoiesis, upregulated CODEGs were constantly enriched in stem cells and progenitors, whereas downregulated CODEGs were enriched in committed cells. This finding suggests that, as in normal differentiation, a gradient of gene expression still exists throughout AML hierarchy and that LSCs maintain the expression of a pool of common genes involved in HSC maintenance and proliferation.
The unbiased CODEG22 score was independent of current prognostic factors: age, cytogenetics, and molecular abnormalities. Although multiple AML gene expression signatures have been established during the past 2 decades, Ng et al recently proposed a simple model, called LSC17,12 which outperformed previous prognostic scores in the stratification of adult11,13,55 and pediatric AML.12,56 They proposed that the strength of the LSC17 model comes from the biological properties of LSCs, which may confer resistance to therapy. However, the absence of the favorable cytogenetic group in their data set could have affected the initial LSC gene list used to generate the LSC17 score. Indeed, LSC17 was recently reported to perform poorly on the favorable subgroup in pediatric AML.56 Likewise, our score could not identify poor responders within the favorable subtype, despite taking t(8;21) and t(9;11) translocations into account while identifying CODEGs. This result suggests that the mechanisms of relapse in the favorable subgroups, especially for samples harboring CBF mutations, may be different from those of the other subgroups and require special attention. In addition, it has been reported that Ara-C resistance and relapse could also result from the evolution of non-LSC populations.16,17 The CODEG22 model is based on a mixed signature that combines genes related to both stemness (4 genes) and myeloid differentiation (18 genes). The result of the comparison with the LSC17 signature demonstrated that the prognostic power of the CODEG22 score comes, at least partially, from blast-enriched genes, indicating that molecular markers of differentiation in AML contain valuable prognostic information that should not be underestimated in the prediction of a patient’s survival. Indeed, it has been reported that the mechanisms shaping drug tolerance and the subsequent relapse in acute leukemia can be present in relapse-fated cells at diagnosis.57,58 Seven upregulated CODEGs were also increased at relapse, suggesting that cells expressing these genes are selected by chemotherapy or that chemotherapy can induce their expression in relapse-fated cells. We speculate that the prognostic power of CODEG22 score could be associated with mechanisms related to drug resistance in relapse-fated cells at diagnosis.
Of interest, the CODEG22 score stratified patients within the intermediate- and adverse-risk group in the training cohort and also stratified cytogenetically abnormal AML (CA-AML) and CN-AML subgroups, as well as elderly patients from independent data sets. This finding suggests that CODEG22 may complement ELN classification for more accurate prediction of the disease outcome, independent of NPM1 or FLT3 status. The accuracy of prognostic models in AML can benefit from incorporating multiple data types, including gene expression data.59 Indeed, our results indicate that CODEG22 can improve stratification of patients when combined with ELN classification. We also validated our results through a retrospective model using real-time qPCR. The expression of signature genes in clinical routine can be easily evaluated by real-time qPCR. A score can be calculated for each patient by using the weighted sum of signature gene expression for use in clinical practice. Nevertheless, further prospective validation, both outside and within clinical trials, is necessary to confirm these findings.
In summary, using robust analysis of differential gene expression, we identified a common set of DEGs across AML cytogenetic subgroups, which included crucial genes that are worth testing as targets in future treatments. Ara-C combined with an anthracycline remains the worldwide standard for the treatment of young patients with AML.60 Hence, risk stratification of patients remains the main option for defining treatment intensity and deciding whether to perform allogeneic stem cell transplantation. We created a prognostic model of AML using differentiation markers, which proved to be robust and unbiased and could complement the ELN classification for more accurate prediction of clinical outcomes.
The normalized data set of 1732 integrated samples reported in this article is available on the Gene Expression Omnibus (GEO) database (accession number GSE147515).
Acknowledgments
The authors thank K. Kranc, O. Bernard, and F. Pflumio for valuable discussions and review of the manuscript, and Lamya Haddaoui for obtaining samples from the GOELAMSthèque/FILOthèque.
This work was supported by a grant from “Fondation de France” (A.N. and F.M.); a grant from Fondation Association pour la Recherche sur le Cancer (ARC) (H.D.); funding from the French Committees of the “Ligue Contre le Cancer Grand-Ouest” (16 [Charente], 37 [Indre-et-Loire], and 86 [Vendée]) (F.M.), the Hubert Curien Program (CEDRE) (F.M. and K.Z.); the Lebanese University (K.Z.); and the Lebanese National Council for Scientific Research (K.Z.).
Authorship
Contribution: A.N., H. Dakik, K.Z., and F.M. designed the study; A.N. downloaded and assembled the data set; A.N., H. Dakik, and F.P. acquired the data; A.N., H. Dakik, O.H., K.Z. and F.M. analyzed the data; K.Z. and F.M. supervised the work; E.G., A.P., C.R., and M.C.B. provided samples from the French GOELAMSthèque/FILOthèque; M.C., C.P., H. Dombret, and J.L. provided data from the French ALFA-0701 cohort; A.N., H. Dakik, K.Z., and F.M. wrote the manuscript; and all authors reviewed and edited the manuscript.
Conflicts-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Frédéric Mazurier, CNRS ERL7001 LNOx, EA 7501 University of Tours, 10 Tonnellé Blvd, BP 3223, 37032 Tours Cedex 01, France; e-mail: frederic.mazurier@inserm.fr; Ali Nehme, McGill University and Genome Quebec Innovation Centre, 740 Dr Penfield St, Montreal, QC H3T1E6, Canada; e-mail: ali.nehme2@mcgilll.ca; and Hassan Dakik, McGill University, RI-MUHC, Glen Site, 1001 Décarie Blvd, Montreal, QC H4A3J1, Canada; e-mail: hassan.dakik@mail.mcgill.ca.