

Key Points
Knockin of human VWF exon 28 and crossbreeding with hGPIbαTg generated a humanized mouse model of platelet GPIbα-VWFA1 interaction.
A humanized GPIbα-VWFA1 mouse model is useful for studying human GPIbα-VWF function and for developing antithrombotic intervention.

Abstract
The interaction of platelet glycoprotein Ibα (GPIbα) with von Willebrand factor (VWF) initiates hemostasis after vascular injury and also contributes to pathological thrombosis. GPIbα binding to the VWF A1 domain (VWFA1) is a target for antithrombotic intervention, but attempts to develop pharmacologic inhibitors have been hindered by the lack of animal models because of the species specificity of the interaction. To address this problem, we generated a knockin mouse with Vwf exon 28–encoding domains A1 and A2 replaced by the human homolog (VWFh28). VWFh28 mice (M1HA) were crossbred with a transgenic mouse strain expressing human GPIbα on platelets (mGPIbαnull;hGPIbαTg; H1MA) to generate a new strain (H1HA) with humanized GPIbα-VWFA1 binding. Plasma VWF levels in the latter 3 strains were similar to those of wild-type mice (M1MA). Compared with the strains that had homospecific GPIbα-VWF pairing (M1MA and H1HA), M1HA mice of those with heterospecific pairing had a markedly greater prolongation of tail bleeding time and attenuation of thrombogenesis after injury to the carotid artery than H1MA mice. Measurements of GPIbα-VWFA1 binding affinity by surface plasmon resonance agreed with the extent of observed functional defects. Ristocetin-induced platelet aggregation was similar in H1HA mouse and human platelet-rich plasma, and it was comparably inhibited by monoclonal antibody NMC-4, which is known to block human GPIbα-VWFA1 binding, which also inhibited FeCl3-induced mouse carotid artery thrombosis. Thus, the H1HA mouse strain is a fully humanized model of platelet GPIbα-VWFA1 binding that provides mechanistic and pharmacologic information relevant to human hemostatic and thrombotic disorders.
Introduction
Arterial thrombosis that causes myocardial infarction and stroke is a major health problem1 in which platelets play a central role.2-4 Antiplatelet agents, mainly aspirin5 and P2Y12 receptor antagonists,6 along with GPIIb-IIIa (αIIbβ3) inhibitors,7 are used for prevention and treatment of thrombosis. However, recurrent thrombosis is a persistent problem8 ; thus, developing more efficient antithrombotic agents that minimally interfere with hemostasis remains an important goal. At sites of vascular injury, initial platelet adhesion to the subendothelium is mediated by the von Willebrand factor (VWF) A1 domain (VWFA1) binding to glycoprotein Ibα (GPIbα) in the platelet membrane GPIb-IX-V receptor complex.9,10 This interaction, which is critical for thrombus formation under high shear stress in rapidly flowing blood, is a target for the development of antithrombotic agents.11,12 Anucleated mammalian platelets and nucleated thrombocytes in other vertebrates all use GPIbα to bind VWF, but the species specificity of the interaction complicates the use of animal models for evaluating candidate pharmacologic antagonists. Notably, mouse GPIbα interacts poorly with human VWF,13 whereas human GPIbα binds endogenous mouse VWF well enough to support a normal tail bleeding time.14,15
To overcome the mouse-human GPIbα-VWFA1 compatibility barrier, in previous studies, mouse VWF was mutated to allow binding to transfused human platelets,16 or chimeric human VWF with mouse A1 domain was transiently expressed by hydrodynamic injection to target VWF interactions other than that with GPIbα.17 These efforts left unsolved the problem of generating a mouse model with humanized GPIbα-VWFA1 binding. Here, we report the results of a knockin strategy to replace Vwf exon 28 (encoding domains A1 and A2) with the human homolog. In the resulting mouse strain (M1HA), which expresses native GPIbα and chimeric VWF with human A1 domain under the control of the respective promoters, VWF is synthetized by endothelial cells and megakaryocytes. Crossbreeding with mouse GPIbαnull (mGPIbαnull) mice transgenically expressing human GPIbα resulted in a fully humanized mouse model of GPIbα-VWF binding (H1HA). Thrombogenesis tests in the new M1HA and H1HA strains compared with wild-type (WT) M1MA and H1MA (expressing human GPIbα with endogenous mouse VWF)15 reflected the different affinities of the 4 possible human and mouse GPIbα-VWFA1 combinations measured with purified proteins. Thus, the H1HA mice can be used to understand structure-function relationships in GPIbα-VWF binding and evaluate candidate inhibitory drugs more directly relevant to the treatment of human hemostatic and thrombotic disorders.
Materials and methods
Surface plasmon resonance
To analyze the GPIbα-VWF interaction by surface plasmon resonance (SPR; Biacore 3000, GE), we generated a human or mouse recombinant amino-terminal GPIbα fragment (residues 2-290 or 2-283, respectively) followed by 133 residues of the SV40 large T antigen with Cys residues mediating dimerization and a monoclonal antibody (LJ-3A2) specific for the SV40 sequence.18 After linking to an SPR chip (HC200M, XanTec Bioanalytics), the latter immobilized GPIb fragments in the appropriate orientation to measure kinetics parameters of VWFA1 binding. Varying concentrations (0.78-100 nM) of the recombinant dimeric VWFA1 fragment with human or mouse sequences (VWF subunit residues 445-733 or 445-716, respectively [add 763 for the number in pre-pro-VWF]) were prepared in 135 mM NaCl, 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES-buffered saline, pH 7.4) containing 0.005% Tween 20. VWFA1 solutions were injected over the chip (association phase) at 75 μL/min for 3 minutes followed by solution buffer for 20 minutes (dissociation phase). Dissociation data points were used to obtain an off-rate constant (Koff) by nonlinear regression of a one-phase exponential decay model. The Koff was then used as a constraint to compute Kon and KD (Koff:Kon ratio) according to a Langmuir model of interaction. Recombinant GPIbα amino-terminal and dimeric VWFA1 fragments were expressed in Drosophila melanogaster S2 cells and purified from culture supernatant as described previously.18,19
Generation of human VWF exon 28 knockin mice (VWFh28) and crossbreeding with human GPIbα transgenic mice
The VWFh28 targeting vector containing human VWF exon 28, neomycin selection cassette, loxP recombinase target sequences, and a thymidine kinase gene for negative selection was electroporated into E14 Tg2a.4 embryonic stem (ES) cells. To ensure correct splicing in mouse genomic DNA, the 5′ and 3′ ends of exon 28 were kept as mouse nucleotides, which resulted in replacement of residues 471 to 904 (1234-1667 in pre-pro-VWF) with the human VWF sequence. After selection by continuous growth in the presence of neomycin and ganciclovir, 380 clones were screened by polymerase chain reaction (PCR) and those that were positive were confirmed by Southern blot. Correctly targeted ES cells were injected into blastocyst stage embryos to generate chimeric mice. To remove the loxP-flanked Neo cassette, these mice were bred with B6.FVB-Tg (EIIa-Cre) C5379Lmgd/J mice (The Jackson Laboratory) constitutively expressing Cre recombinase. VWFh28 mice (M1HA; Table 1) were then crossed with the human GPIbα transgenic mouse strain (mGPIbαnull/hGPIbαTg [H1MA]) in which platelets expressed only human GPIbα in the GPIb-IX-V complex, thus generating mice with humanized GPIbα-VWF interaction (H1HA). To accelerate backcrossing, transgene-positive mice with the highest percentage of C57BL/6J strain-specific genomic polymorphisms were used as breeders for successive generations (Marker-Assisted Accelerated Backcrossing; Max-Bax, Charles River).
Nomenclature of mouse strains with different mouse and human GPIbα-VWF exon 228 (A1 and A2 domains) combinations
| M1MA | M1HA | H1MA | H1HA | KO1HA | H1KOA | |
|---|---|---|---|---|---|---|
| GPIbα | Mouse | Mouse | Human | Human | Null | Human |
| VWFA1 and VWFA2 | Mouse | Human | Mouse | Human | Human | Null |
| M1MA | M1HA | H1MA | H1HA | KO1HA | H1KOA | |
|---|---|---|---|---|---|---|
| GPIbα | Mouse | Mouse | Human | Human | Null | Human |
| VWFA1 and VWFA2 | Mouse | Human | Mouse | Human | Human | Null |
Tail bleeding time
Mice were anesthetized using isoflurane in a precision vaporizer. After clipping 3 mm of the distal tip of the tail with a sterile scalpel blade, the tail was immersed in isotonic saline at 37°C, and the time to complete cessation of blood flow was recorded as the bleeding time.20,21 After 600 seconds, persistent hemorrhage was stopped by cauterizing the tail wound. All protocols for animal studies were approved by the Institutional Animal Care and Use Committees of The Scripps Research Institute and the Medical College of Wisconsin.
In vivo arterial thrombosis
The common carotid artery of anesthetized mice was dissected, and an ultrasound flow probe was positioned around the vessel.22 After measuring baseline flow, a 10.8-μL drop of 8% (0.30 M) or 9% (0.34 M) FeCl3⋅6H2O23 was applied to the surface of the adventitia for 3 minutes. After the strip of filter paper was removed and the surrounding area was washed, carotid blood flow was monitored for the total observation period of 33 minutes. The artery was considered occluded when flow was <0.1 mL/min. Stable occlusion was defined as flow rate <0.1 mL/min for at least 10 minutes. A flow index was calculated as the ratio between the volume of blood that flowed through the carotid artery during the observation time and the volume calculated, assuming a constant flow rate equal to the maximum value observed during the first minute after injury.
Plasma VWF concentration, multimer distribution, and factor VIII activity assay
VWF antigen was measured by enzyme-linked immunosorbent assay21 using a rabbit anti-human VWF polyclonal antibody (immunoglobulin G) cross-reacting with mouse VWF (produced at The Scripps Research Institute). Normal pooled plasma from C57BL/6J WT mice was used as a reference and defined as 1 U/mL. VWF multimers were analyzed by electrophoresis through 1% HGT(P) agarose containing 0.1% sodium dodecyl sulfate followed by western blotting with the same antibodies as those used for enzyme-linked immunosorbent assay. Factor VIII (FVIII) activity was tested by using a thrombin generation assay with human FVIII-deficient plasma and tissue factor/FIXa (0.15 pM and 200 pM, respectively) with CaCl2 (18 mM) to initiate coagulation, as previously described.23
Mouse platelet analyses
Blood obtained by retro-orbital bleeding into sodium citrate anticoagulant (10.88 mM final) was diluted (1:1) with Tyrode’s buffer (pH 7.4). Platelet-rich plasma (PRP) was prepared by centrifugation at 230g for 7 minutes at room temperature. Ristocetin-induced platelet aggregation was performed with platelets at 3 × 1011/L at 37°C using a Model 440 aggregometer (Chrono-Log Corporation). Complete blood counts were obtained with the IDEXX ProCyte DX analyzer (IDEXX Laboratories Inc.).
PCR
Genomic DNA prepared from leukocytes with the QIAamp DNA Blood Mini Kit (Qiagen) was analyzed by PCR with forward primer specific for human or mouse VWF exon 28 (5′-CCCTGAGAACAAGGCCTTCG-3′ or 5′-GGAACTGGAGAGGATCAGCA-3′, respectively) in combination with a common mouse intron 28 reverse primer (5′-TTAGCAAAGGCCAAAATTATA-3′). Amplification was performed using GoTaq Green Master Mix (Promega).
Molecular dynamics simulations
The role of distinct residues at the interface of GPIbα-VWFA1 complexes was analyzed by molecular dynamics (MD) simulations. The average displacement of backbone atoms in each frame relative to the reference frame (which indicates the global stability of each complex) was measured as root mean square deviation (RMSD). The fluctuation of backbone atoms relative to their average position during the simulation (which identifies residues in regions with high flexibility) was measured as root mean square fluctuation (RMSF). Stabilization of RMSD values over time was used to identify the time when complexes reached conformational equilibrium (starting from 100 ns) from which the RMSF was calculated. More details are provided in the supplemental Data.
Results
Cross-species specificity of human and mouse GPIbα-VWFA1 binding
As previously shown, human GPIbα rescues the bleeding phenotype of GPIbαnull mice,15 but mouse platelet adhesion to human VWF is markedly reduced.13 To further evaluate the apparently variable cross-species compatibility in GPIbα-VWF binding, we analyzed soluble VWFA1 domain binding to an immobilized GPIbα amino-terminal fragment by SPR. Human VWFA1 binds to mouse GPIbα with 54 times lower affinity than mouse VWFA1 (KD = 39.64 vs 0.74 nM), but mouse VWFA1 binding to human GPIbα was much more efficient, with only 2.8 times lower affinity than human VWFA1 (KD = 4.19 vs 1.48 nM). In both cases, the reduced affinity resulted mainly from a decreased association rate, which was 19 times slower for human than for mouse VWFA1 binding to mouse GPIbα but only 2.1 times slower for mouse than for human VWFA1 binding to human GPIbα (Table 2). (Sensograms of representative experiments are shown in supplemental Figure 1.) To evaluate the functional consequences of these differences in vivo, we generated 2 new mouse strains expressing human A1 or A2 domains within mouse VWF in the context of either mouse or human GPIbα, the latter resulting in a fully humanized GPIbα-VWFA1 interaction.
Parameters of mouse and human GPIbα-VWFA1 interaction
| GPIbα | VWFA1 | KD (nM) | Kon (M-1s-1) | Koff (s-1) |
|---|---|---|---|---|
| Mouse | Mouse | 0.74 ± 0.10 | 3.52 ± 0.45 × 106 | 2.62 ± 0.47 × 10-3 |
| Mouse | Human | 39.64 ± 10.59 | 0.19 ± 0.01 × 106 | 7.49 ± 1.92 × 10-3 |
| Human | Mouse | 4.19 ± 1.83 | 1.51 ± 0.87 × 106 | 5.30 ± 1.46 × 10-3 |
| Human | Human | 1.48 ± 0.45 | 3.11 ± 0.83 × 106 | 4.77 ± 2.23 × 10-3 |
| GPIbα | VWFA1 | KD (nM) | Kon (M-1s-1) | Koff (s-1) |
|---|---|---|---|---|
| Mouse | Mouse | 0.74 ± 0.10 | 3.52 ± 0.45 × 106 | 2.62 ± 0.47 × 10-3 |
| Mouse | Human | 39.64 ± 10.59 | 0.19 ± 0.01 × 106 | 7.49 ± 1.92 × 10-3 |
| Human | Mouse | 4.19 ± 1.83 | 1.51 ± 0.87 × 106 | 5.30 ± 1.46 × 10-3 |
| Human | Human | 1.48 ± 0.45 | 3.11 ± 0.83 × 106 | 4.77 ± 2.23 × 10-3 |
Binding parameters were measured by SPR using surface-immobilized GPIbα amino-terminal fragment and soluble dimeric VWFA1.
Generation of mice with chimeric human-mouse VWF by knockin of human VWF exon 28
Because of the difficulty of inserting the large human VWF gene (178 kb) into the mouse Vwf locus,24 we replaced mouse Vwf exon 28 with the human homolog (residues 471-904 or 1234-1667 in pre-pro-VWF), which similarly encodes domains A1 and A2. To implement this strategy, mouse ES cells were targeted with a vector containing human VWF exon 28 with 5′ and 3′ flanking homology arms (Figure 1A). Correctly targeted clones identified by PCR screening and Southern blot (Figure 1B) were used for blastocyst injection to produce chimeric mice. VWFh28 knockin mice were crossed with EIIa-Cre transgenic mice to delete the floxed Neo cassette and then bred to homozygosity. Mice expressing chimeric VWF with human A1 domain (VWFh28) and mouse GPIbα (M1HA) were crossbred with transgenic mice expressing only human GPIbα on platelets (H1MA)14,15 to obtain animals expressing human GPIbα and VWFh28 (H1HA). All the mouse strains used to evaluate the species specificity of the GPIbα-VWF interaction are listed in Table 1.
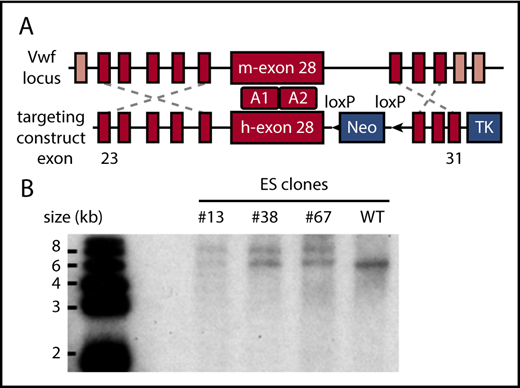
Establishing the VWFh28knockin mouse strain. (A) E14 mouse ES cells were targeted with the depicted construct, and a total of 380 clones were screened by PCR. (B) Genomic DNA samples of the positive ES clones were digested with restriction enzyme KpnI and confirmed by Southern blot using a probe designed in the 3′ homology arm. The WT allele appears as a 6.2-kb band whereas the targeted allele appears as a 7.8-kb band. TK, thymidine kinase gene.
Establishing the VWFh28knockin mouse strain. (A) E14 mouse ES cells were targeted with the depicted construct, and a total of 380 clones were screened by PCR. (B) Genomic DNA samples of the positive ES clones were digested with restriction enzyme KpnI and confirmed by Southern blot using a probe designed in the 3′ homology arm. The WT allele appears as a 6.2-kb band whereas the targeted allele appears as a 7.8-kb band. TK, thymidine kinase gene.
Values of selected blood parameters in mice expressing VWFh28 and human GPIbα
Plasma VWF levels and multimer profiles as well as blood platelet counts were comparable in mice expressing endogenous mouse VWF or chimeric VWFh28 with either mouse (WT M1MA and M1HA) or human (H1MA and H1HA) GPIbα (Figure 2A-C). For unknown reasons, only the procoagulant activity of FVIII, which binds to VWF domains D′ to D3, was modestly but significantly lower in M1HA than in WT M1MA mice (Figure 2D).
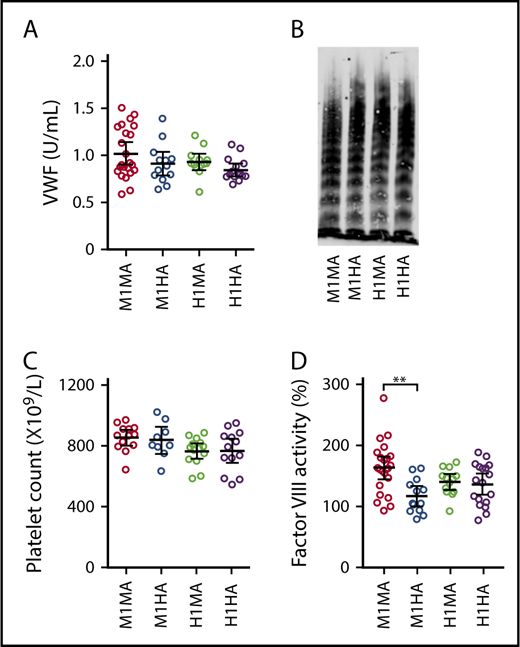
Selected parameters relevant to hemostasis and thrombogenesis in the mouse strains used for this study. (A) Plasma concentration of VWFh28 (M1HA and H1HA) compared with endogenous mouse VWF (M1MA and H1MA) measured by enzyme-linked immunosorbent assay (ELISA) using anti-human VWF polyclonal antibodies. M1MA, n = 23; M1HA, n = 13; H1MA, n = 13; H1HA, n = 15. (B) Multimer analysis of mouse plasma samples showing the presence of full-range multimers in all 4 tested mouse strains. (C) Platelet counts. (D) FVIII activity of mouse plasma serially diluted with human FVIII-deficient plasma and measured in a thrombin generation test initiated by 0.15 pM tissue factor/200 pM FIXa, and CaCl2. Values are percent of normal human reference plasma. All numerical data are shown with mean ± 95% confidence intervals. Statistical analysis performed with one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test (P = .001405) and Kruskal-Wallis nonparametric test followed by Dunn’s multiple comparison test (P = .002574). **P < .01 where indicated; all other comparisons were not significantly different.
Selected parameters relevant to hemostasis and thrombogenesis in the mouse strains used for this study. (A) Plasma concentration of VWFh28 (M1HA and H1HA) compared with endogenous mouse VWF (M1MA and H1MA) measured by enzyme-linked immunosorbent assay (ELISA) using anti-human VWF polyclonal antibodies. M1MA, n = 23; M1HA, n = 13; H1MA, n = 13; H1HA, n = 15. (B) Multimer analysis of mouse plasma samples showing the presence of full-range multimers in all 4 tested mouse strains. (C) Platelet counts. (D) FVIII activity of mouse plasma serially diluted with human FVIII-deficient plasma and measured in a thrombin generation test initiated by 0.15 pM tissue factor/200 pM FIXa, and CaCl2. Values are percent of normal human reference plasma. All numerical data are shown with mean ± 95% confidence intervals. Statistical analysis performed with one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test (P = .001405) and Kruskal-Wallis nonparametric test followed by Dunn’s multiple comparison test (P = .002574). **P < .01 where indicated; all other comparisons were not significantly different.
Human and mouse species specificity of the GPIbα-VWFA1 interaction and its effect on hemostasis
The tail bleeding time of C57BL/6J WT mice (M1MA) was <2 minutes (Figure 3A)20,21 as opposed to >10 minutes in mice expressing human GPIbα or VWFh28 but lacking VWF (H1KOA) or GPIbα (KO1HA), respectively. The bleeding time of mice expressing human GPIbα and endogenous or chimeric human VWFh28 (H1MA and H1HA) was not different from that in WT mice, although in 3 of 17 H1HA mice, the hemorrhage lasted ∼3 minutes. In contrast, mice expressing endogenous GPIbα and human VWFh28 (M1HA) had a longer bleeding time than all other strains except GPIbα and VWF knockout strains (Figure 3A). This result is in agreement with the greatly reduced affinity of human VWFA1 binding to mouse GPIbα measured by SPR as well as the previously reported lack of mouse platelet adhesion to immobilized human VWF.13 Of note, only 3 of 6 M1HA mice with tail bleeding time of >10 minutes had a blood loss volume comparable to that of mice lacking GPIbα or VWF, which both had a 10-minute hemorrhage volume significantly greater than that of all other strains (Figure 3B).

Tail bleeding time in mouse strains with different mouse and human VWF exon 28 and GPIbα combinations. (A) Bleeding time in 6 mouse strains: M1MA, n = 13; M1HA, n = 17; H1MA, n = 12; H1HA, n = 17; KO1HA, n = 4; H1KOA, n = 5 (Table 1). Mouse strains were tested as described in “Materials and methods.” (B) The amount of blood lost into collection tubes was determined by measuring hemoglobin content. The number of mice (n values) are the same as for the bleeding time test, except for M1HA (n = 16). Data are shown with median and interquartile range. Statistical analysis in panel A was performed with the Kruskal-Wallis nonparametric test followed by Dunn’s multiple comparison test; in panel B, one-way ANOVA followed by Tukey’s multiple comparison test was used. *P < .05; **P < .0; ***P < .001.
Tail bleeding time in mouse strains with different mouse and human VWF exon 28 and GPIbα combinations. (A) Bleeding time in 6 mouse strains: M1MA, n = 13; M1HA, n = 17; H1MA, n = 12; H1HA, n = 17; KO1HA, n = 4; H1KOA, n = 5 (Table 1). Mouse strains were tested as described in “Materials and methods.” (B) The amount of blood lost into collection tubes was determined by measuring hemoglobin content. The number of mice (n values) are the same as for the bleeding time test, except for M1HA (n = 16). Data are shown with median and interquartile range. Statistical analysis in panel A was performed with the Kruskal-Wallis nonparametric test followed by Dunn’s multiple comparison test; in panel B, one-way ANOVA followed by Tukey’s multiple comparison test was used. *P < .05; **P < .0; ***P < .001.
Human and mouse species specificity of the GPIbα-VWFA1 interaction and its effect on thrombogenesis
To evaluate how different mouse and human GPIbα-VWFA1 combinations influenced endoarterial thrombus formation, we measured time to occlusion and flow index22 in the FeCl3 carotid artery injury model.25 After a lesion was induced by 8% FeCl3, occlusion developed in all WT (M1MA) mice, and all mice but 1 were stable, although there was no occlusion in M1HA mice and only 3 transient occlusions in H1MA mice; the latter strains had a significantly higher flow index than WT mice (Figure 4A). H1HA mice developed 6 occlusions (67%) of which 3 (33%) became stable. The parameters of time to stable occlusion as well as the flow index in H1HA mice differed significantly from those in WT M1MA mice (Figure 4A), indicating a decreased thrombogenic potential in the homospecific human compared with mouse GPIbα-VWFA1 interaction. After a more severe lesion induced by 9% FeCl3, stable occlusion with a similar flow index developed in all M1MA and H1HA mice, whereas M1HA mice had a significantly higher flow index than all other strains (Figure 4B). Occlusion frequency was not different in M1MA, H1HA, and H1MA mice, although the occlusion was not stable in 2 of 7 in the H1MA group, and the flow index was higher in those animals (Figure 4B).
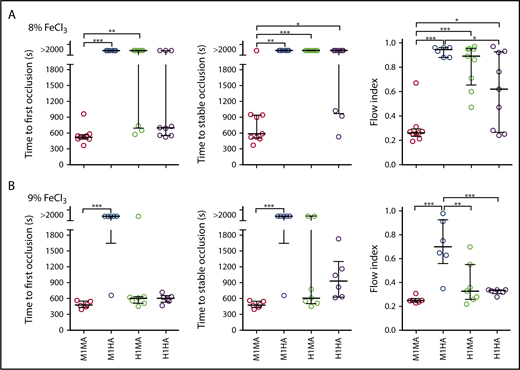
FeCl3-induced carotid artery thrombosis model. The common carotid artery of anesthetized mice was exposed to (A) 8% or (B) 9% FeCl3, and blood flow was monitored for 30 minutes afterward. Time to first occlusion is defined as time recorded until the blood flow rate decreased to <0.1 mL/min; time to stable occlusion is defined as time until flow <0.1 mL/min was recorded for at least 10 minutes. Flow index is the ratio between the recorded blood volume flowing through the artery in 30 minutes and that calculated assuming a constant flow at the maximum rate recorded during the first minute after injury. Arteries that did not occlude were reported as time to first and stable occlusion >2000 seconds. (A) M1MA, n = 9; M1HA, n = 6; H1MA, n = 9; H1HA, n = 9. (B) M1MA, n = 6; M1HA, n = 6; H1MA, n = 7; H1HA, n = 6. Data are shown with median and interquartile range. Statistical analysis was performed with the Kruskal-Wallis nonparametric test followed by Dunn’s multiple comparison test for first and stable occlusion and by ordinary one-way ANOVA followed by Tukey’s multiple comparison test for flow index. *P < .05; **P < .01; ***P < .001.
FeCl3-induced carotid artery thrombosis model. The common carotid artery of anesthetized mice was exposed to (A) 8% or (B) 9% FeCl3, and blood flow was monitored for 30 minutes afterward. Time to first occlusion is defined as time recorded until the blood flow rate decreased to <0.1 mL/min; time to stable occlusion is defined as time until flow <0.1 mL/min was recorded for at least 10 minutes. Flow index is the ratio between the recorded blood volume flowing through the artery in 30 minutes and that calculated assuming a constant flow at the maximum rate recorded during the first minute after injury. Arteries that did not occlude were reported as time to first and stable occlusion >2000 seconds. (A) M1MA, n = 9; M1HA, n = 6; H1MA, n = 9; H1HA, n = 9. (B) M1MA, n = 6; M1HA, n = 6; H1MA, n = 7; H1HA, n = 6. Data are shown with median and interquartile range. Statistical analysis was performed with the Kruskal-Wallis nonparametric test followed by Dunn’s multiple comparison test for first and stable occlusion and by ordinary one-way ANOVA followed by Tukey’s multiple comparison test for flow index. *P < .05; **P < .01; ***P < .001.
To further characterize the humanized GPIbα-VWF interaction in mice, we tested the response of PRP to ristocetin to which mouse PRP is not responsive.14 Of the 4 strains tested, only PRP from H1HA mice showed dose-dependent ristocetin-induced aggregation that was completely inhibited by the anti-human VWFA1 mouse monoclonal antibody NMC-426-28 (Figure 5A-C). Injected in vivo, the monovalent NMC-4 Fab fragment prolonged the tail bleeding time to >10 minutes (Figure 5D) and completely prevented 9% FeCl3-induced carotid artery thrombosis (Figure 5E).
![Figure 5. In vitro and in vivo effects of an anti-human VWFA1 monoclonal antibody in the H1HA mouse strain. (A) Response to ristocetin (1.5 mg/mL) in PRP from 4 mouse strains; platelet aggregation is evident only in H1HA mice. (B) Dose-response of H1HA PRP to the indicated ristocetin concentrations. (C) The anti-human VWFA1 monoclonal antibody NMC-4 (5 μg/mL; 1 minute incubation), but not 5 μg/mL of nonimmune control immunoglobulin G (IgG), completely blocked ristocetin-induced platelet aggregation in H1HA mouse PRP. (D) Treatment of H1HA mice with NMC-4 (0.8 mg/kg injected IV 7 minutes before the start of the test procedure [n = 6]) prolonged the tail bleeding time to >10 minutes as compared with <3 minutes in mice treated with control IgG (n = 6). (E) The same dose of NMC-4 prevented carotid artery occlusion in H1HA mice after a 9% FeCl3–induced lesion (n = 10 for both treated and control mice). (A-C) Platelet aggregation is shown as a time-dependent increase in light transmittance through PRP. (D-E) Individual data are shown with median and interquartile range. Statistical analysis was performed with the Mann-Whitney nonparametric U test. **P < .01; ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/2/19/10.1182_bloodadvances.2018023507/7/m_advances023507f5.png?Expires=1769123537&Signature=KKWwUVyJLUL6tv-FCxxdWuDbmoyOh9Oh0bLwwZEtoGKlaNrxebxSX9x~5XrRg27dGf8c62HyanxUACpNnggZCWyBvg6CTHsLiRb~67tjFoKH7W2bz8ANBMeGJsASlp6eL5vXz7hKuqWrfVcSuccCL201kvAC~DVX-mJZk~yrJu34~psIZxNDraj5HMAUezytoJGCFAgACyO4kJHUduVCBS1Jo6z1TAC5~pqcKjmpwX01pIhf5EbjPAUarAflAHtBsibpJSSAXu63hEAR0eNb7emOLMT9qKeY84nA2~-k2Zf8ux00ccYZ71j0raXGIlmOHabtbdMimcmLeojnAOSx2g__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
In vitro and in vivo effects of an anti-human VWFA1 monoclonal antibody in the H1HA mouse strain. (A) Response to ristocetin (1.5 mg/mL) in PRP from 4 mouse strains; platelet aggregation is evident only in H1HA mice. (B) Dose-response of H1HA PRP to the indicated ristocetin concentrations. (C) The anti-human VWFA1 monoclonal antibody NMC-4 (5 μg/mL; 1 minute incubation), but not 5 μg/mL of nonimmune control immunoglobulin G (IgG), completely blocked ristocetin-induced platelet aggregation in H1HA mouse PRP. (D) Treatment of H1HA mice with NMC-4 (0.8 mg/kg injected IV 7 minutes before the start of the test procedure [n = 6]) prolonged the tail bleeding time to >10 minutes as compared with <3 minutes in mice treated with control IgG (n = 6). (E) The same dose of NMC-4 prevented carotid artery occlusion in H1HA mice after a 9% FeCl3–induced lesion (n = 10 for both treated and control mice). (A-C) Platelet aggregation is shown as a time-dependent increase in light transmittance through PRP. (D-E) Individual data are shown with median and interquartile range. Statistical analysis was performed with the Mann-Whitney nonparametric U test. **P < .01; ***P < .001.
In vitro and in vivo effects of an anti-human VWFA1 monoclonal antibody in the H1HA mouse strain. (A) Response to ristocetin (1.5 mg/mL) in PRP from 4 mouse strains; platelet aggregation is evident only in H1HA mice. (B) Dose-response of H1HA PRP to the indicated ristocetin concentrations. (C) The anti-human VWFA1 monoclonal antibody NMC-4 (5 μg/mL; 1 minute incubation), but not 5 μg/mL of nonimmune control immunoglobulin G (IgG), completely blocked ristocetin-induced platelet aggregation in H1HA mouse PRP. (D) Treatment of H1HA mice with NMC-4 (0.8 mg/kg injected IV 7 minutes before the start of the test procedure [n = 6]) prolonged the tail bleeding time to >10 minutes as compared with <3 minutes in mice treated with control IgG (n = 6). (E) The same dose of NMC-4 prevented carotid artery occlusion in H1HA mice after a 9% FeCl3–induced lesion (n = 10 for both treated and control mice). (A-C) Platelet aggregation is shown as a time-dependent increase in light transmittance through PRP. (D-E) Individual data are shown with median and interquartile range. Statistical analysis was performed with the Mann-Whitney nonparametric U test. **P < .01; ***P < .001.
Structural dynamics of the GPIbα-VWFA1 complexes
To ascertain how structural stability of the 4 possible mouse/human (M/H) interspecies VWFA1 (A)-GPIbα (1) complexes (M1MA, M1HA, H1MA, and H1HA) contributed to the observed results, RMSD and RMSF were computed from the MD trajectories. Trajectory analysis revealed an initial increase of backbone RMSD until equilibrium was reached (100 ns) for all the complexes; then, no further global variations were found (supplemental Figure 3). Similarly, RMSF analysis showed no significant differences for VWFA1, including residues at the interface with the β-hairpin of GPIbα (Arg57 in mice; His57 in humans) with RMSF values smaller than 1 Å, showing that this region is stable (Figure 6A-E). For GPIbα, RMSF analysis showed even fewer differences among complexes compared with VWFA1 (Figure 6A-E), except in the β-hairpin region (residues 231-237) of human GPIbα in the H1MA complex, where slightly higher local fluctuations were present (Figure 6C,E). A detailed description of the analysis for each complex is presented in supplemental Data.

Structural stability analysis from MD simulations. (A-D) Backbone RMSF per residue mapped on the structure of each GPIbα-VWFA1 complex: (A) mouse/mouse (M1MA), (B) mouse/human (M1HA), (C) human/mouse (H1MA), and (D) human/human (H1HA) of GPIbα-VWFA1. Structures were colored according to their RMSF values, with a rainbow spectrum ranging from blue (minimum value, 0.30 Å), to red (maximum value, 2 Å). (E) RMSFs along the protein sequences in the complexes; dashed lines highlight hairpin interface regions; secondary structure features are represented by blue (helices) and yellow (β-sheets) boxes along the sequences.
Structural stability analysis from MD simulations. (A-D) Backbone RMSF per residue mapped on the structure of each GPIbα-VWFA1 complex: (A) mouse/mouse (M1MA), (B) mouse/human (M1HA), (C) human/mouse (H1MA), and (D) human/human (H1HA) of GPIbα-VWFA1. Structures were colored according to their RMSF values, with a rainbow spectrum ranging from blue (minimum value, 0.30 Å), to red (maximum value, 2 Å). (E) RMSFs along the protein sequences in the complexes; dashed lines highlight hairpin interface regions; secondary structure features are represented by blue (helices) and yellow (β-sheets) boxes along the sequences.
Discussion
We have analyzed the specificity of human and mouse GPIbα and VWFA1 functions and determined cross-species compatibility of GPIbα-VWFA1 interaction in vitro and in vivo. Compared with homospecific M1MA and H1HA controls, thrombogenesis was impaired in mismatched H1MA and M1HA mice, but the defect was much more pronounced in the latter. M1HA mice exhibited prolonged bleeding and markedly reduced carotid artery thrombosis. These results may reflect the important role of GPIbα-VWFA1 binding under high shear stress when a growing thrombus alters blood flow velocity in the arterial lumen.29-31 This interaction may have less importance in controlling bleeding from small arteries.32 Blood loss volume was smaller with comparably prolonged bleeding in most M1HA mice compared with GPIbα-knockout or VWF-knockout mice.15,33 Thus, as was already known, decreased platelet count and procoagulant activity in the absence of GPIb-IX34 and/or low procoagulant FVIII in the absence of VWF33 also contribute to bleeding. Of note, thrombogenesis after an arterial injury with a lower concentration of FeCl3 (8%) was greater with homospecific mouse than with human GPIbα-VWFA1 pairing, possibly reflecting the lower KD of the former measured by SPR (Table 2). Alternatively, expression of human GPIbα instead of mouse GPIbα may alter binding to ligands other than VWF such as leukocyte Mac-1, which may influence thrombogenesis.35 A potential concern for VWF exon 28 replacement arises as a result of encoding of both A1 (GPIb-binding) and A2 (ADAMTS13 cleavage) domains within exon 28. Species incompatibility between mouse ADAMTS13 and the human VWFA2 domain cleavage target could potentially result in thrombotic thrombocytopenic purpura (TTP) as a result of the persistence of ultra-large VWF multimers. Phenotypic analyses of M1HA and H1HA mice showed VWF multimer profiles as well as platelet counts comparable to those of M1MA mice, which do not implicate TTP. However, it is also known that genetic ablation of Adamts13 alone does not lead to a TTP phenotype in mice, and additional triggers such as a Shiga toxin challenge were required to develop TTP.36,37 Thus, there remains the possibility that a TTP-like phenotype could appear in M1HA and H1HA mice when they are subjected to stress conditions such as bacterial infection, and this point needs to be carefully evaluated.
A previous study found that human and mouse platelet adhesion to mismatched VWFA1 was markedly and similarly decreased when compared with species-matched conditions.16 On the basis of a structural model of the homospecific mouse interaction (M1MA), it was proposed that a salt bridge between GPIbα Asp238 and VWF Arg563 (1326 in pre-pro-VWF) was critical for complex stability. Indeed, this notion agrees with the prolonged tail bleeding time reported in mice with the humanizing Arg563(1326)His substitution in VWF16 and the severe hemostasis and thrombosis defect in M1HA mice documented here. In the same study, the opposite switch in human VWF (His563(1326)Arg) promoted binding to mouse platelets, which led to the conclusion that the indicated salt bridge contributes to mouse GPIbα-VWFA1 species specificity. However, no salt bridge can form between human GPIbα (in which Ala replaces Asp238) and the VWFA1 residue at position 563(1326), whether mouse Arg or human His. Yet, as compared with that in M1MA WT mice, the affinity decreases minimally in H1MA and H1HA mice, and effects on hemostasis and thrombosis are negligible. In contrast, binding of mouse GPIbα with human VWFA1 showed ∼54 times lower affinity (Table 2), and severe functional defects were observed with M1HA mice. These differences cannot be solely explained by lack of the salt bridge between residues 238 in human GPIbα and 563(1326) in VWFA1 (Table 3). It was also emphasized that, in a model of human GPIbα-mouse VWFA1 interaction, mouse VWFA1 Arg563(1326) is not able to efficiently interact with human GPIbα in which Asp238 is replaced by Ala, which leads to an electrostatic clash with K231 (Figure 6).16 However, a Lys-Arg clash cannot readily explain why pairing human instead of mouse GPIbα to mouse VWFA1 results in ∼6 times lower affinity and limited functional consequences, whereas mouse GPIbα with human VWFA1 is functionally incompatible. Therefore, electrostatic perturbations at the contact interface may not contribute significantly to the mouse-human interspecies incompatibility of GPIbα-VWFA1 binding.
Amino acid sequence comparison of the human and mouse GPIbα and VWFA1 fragments used in ex vivo studies
 |
| |
Structurally relevant amino acid residues are indicated by purple boxes. Cysteine residues, all conserved, are indicated by yellow boxes. All other conserved residues are indicated by asterisks. Canonical VWFA1 domain includes residues 497-716 (1260-1479). The portion of domain D3 on the amino-terminal side was expressed to obtain secreted dimers. In the recombinant GPIbα amino-terminal fragment, unpaired Cys65 was mutated to Ala to avoid dimerization after secretion.
In accordance with the latter conclusion, our MD simulations showed comparable stability of homospecific and interspecies GPIbα-VWFA1 complexes and a limited effect of the GPIbα Asp238-VWF Arg563(1326) salt bridge on local structural dynamics. Although the GPIbα β-hairpin shows higher fluctuations in the H1MA interaction, caused by side chain electrostatic clashes, these local perturbations have limited effects on the stability of the complex. Therefore, these structural models, physicochemical SPR data, and functional in vivo hemostasis/thrombosis modes concur to indicate that strong electrostatic interactions are not crucial determinants of complex stability when human GPIbα binds human or mouse VWFA1. MD simulations are not sufficient to address the complex Koff values, which exceed by several orders of magnitude the time scale considered in our models. Nonetheless, the lack of obvious instability factors at this scale suggests that more subtle and slower phenomena are responsible for the affinity differences measured by SPR in agreement with distinct effects on thrombogenesis in vivo. The lack of crystallographic information on the mouse GPIbα amino-terminal domain and heterospecific GPIbα-VWFA1 complexes further limits our interpretations. Moreover, as indicated by SPR data, a dramatic decrease of the association rate constant is the major difference of the greatly dysfunctional M1HA interaction. Such considerations further highlight the limitations on models that mainly test the stability of formed complexes.
Others have attempted to generate mouse models relevant to the screening of candidate pharmacologic inhibitors of VWF-GPIbα binding. One study used hydrodynamic gene transfer to express in vivo human VWF modified to bind mouse platelet GPIb.17 The reported results failed to confirm that switching His563(1326) to Arg allows human VWF to interact with mouse platelets. However, switching 2 stretches of human VWFA1 sequence to mouse residues 563 to 570 (1326-1333) and 607 to 622 (1370-1385), which include His563(1326)Arg and Arg616(1379)His substitutions, changed the species specificity and promoted thrombus formation in mouse blood ex vivo. Surprisingly, the same chimeric VWF did not correct the prolonged tail bleeding time of a VWF-deficient mouse, and only chimeric human VWF with mouse A1 domain replacing the native homolog could restore mouse platelet GPIb-dependent functions in vivo.17 Their results suggest that the entire A1 domain needs to be replaced to convert the species-specific binding property of VWF in vivo. In addition, N-475-508 (1238-1271) and C-696-709 (1459-1472) terminal sequences flanking the A1 domain have been reported as regulatory elements to modulate VWF binding to GPIbα.38 Our VWFh28 strategy replaces the entire A1 domain, including these flanking regions 471-904 (1234-1667), with that of the human sequence, thus creating an ideal model having the humanized property for binding VWFA1 to GPIbα. It should also be noted that transgenic VWF expression by hydrodynamic injection of plasmid DNA, although it yields high plasma levels for some time,39,40 also has limitations. These include ectopic expression, mainly in the liver, bypassing the biosynthetic pathway with regulated VWF storage in endothelial cell Weibel-Palade bodies and megakaryocyte/platelet α-granules, from which VWF is released upon activation and contributes to hemostasis.41 In addition, biosynthesis of VWF requires extensive posttranslational processing, such as glycosylation, which may vary in different cell types.42,43 These concerns may at least partially explain the reason why a higher level of VWF expression in the plasma (300%-500%) was required for the hydrodynamic injection study to correct bleeding time of VWFnull mice.17 Our human VWF exon 28 knockin strategy, in contrast, enabled expression of chimeric VWF under normal physiologic control in endothelial cells, megakaryocytes, and platelets and is expected to serve as an ideal model for studying biological VWF functions in vivo.
Platelet tethering depends on GPIb-VWF interaction at a high shear rate, mostly in the arterial system. Thus, targeting the GPIbα-VWFA1 interaction is expected to have a minor influence on hemostatic capacity in the venous circulation and has been a focus for therapeutic antithrombotic intervention.11 There have been studies testing agents (eg, aptamer and nanobody) that inhibit GPIbα-VWFA1 interaction for their antithrombotic effects.44-46 These agents have not been approved for clinical use for thrombosis, but some of them have been repurposed for testing efficacy in specific pathological conditions such as TTP.47,48 One of the obstacles in developing pharmacologic inhibitors targeting GPIb-VWF is the lack of mouse models because of the species specificity of the interaction. To evaluate GPIbα-VWFA1 inhibitory agents in the mouse system, a rationally designed single-domain inhibitory antibody that recognizes both human and mouse VWFA1 has also been developed.49 Considering the role of GPIb-VWF interaction in primary hemostasis, the risk of bleeding caused by these inhibitors also needs to be carefully evaluated using animal models. Our H1HA mice will be a useful tool for studying the effect of antithrombotic agents and for evaluating the influence on hemostasis and the risk of bleeding.
In conclusion, we have characterized a mouse strain with fully humanized GPIbα-VWFA1 pairing that provides new insights and tools for exploring the mechanisms that control formation and functional regulation of this crucial interaction. These findings should help establish mouse models that better reflect human pathophysiology and thus facilitate the development of drugs targeting GPIbα-VWF50-55 and assessment of their consequences on hemostasis and endovascular thrombosis.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank J. Mattson and C. Boeck at the Blood Research Institute for excellent technical assistance and J.C. Ducom at The Scripps Research Institute High Performance Computing for computational support.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL-56027 and HL-44612 (R.R.M.), HL-117722 and HL-135290 (Z.M.R.), HL-129011 (T.K.), and National Institute of General Medical Sciences grant GM-O69832 (J.E. and S.F.). Additional support was provided by American Heart Association Postdoctoral Fellowship 10POST261016 (S.K.) and by a CSL-Behring Professor Heimburger Award (S.K.). T.K., S.K., and A.Z. were recipients of Junior Faculty Transition Awards from MERU Foundation (Bergamo, Italy).
Authorship
Contribution: S.K. designed and performed experiments, analyzed data, and helped write the manuscript; J.N.O. performed in vivo thrombosis studies; Y.K. performed factor VIII assays; T.K. contributed to experimental design and manuscript writing; Y.M., Y.C., and A.Z. performed experiments and analyzed data; R.S. contributed to designing the targeting vector; S.A.F. contributed to constructing the targeting vector and revised the manuscript; J.E. and S.F. designed the molecular dynamics simulations, and J.E. performed the calculations; S.L.H. contributed to the experimental design and revised the manuscript; and R.R.M. and Z.M.R. designed and directed the study and wrote the manuscript.
Conflict-of-interest disclosure: Z.M.R. is founder, president, and CEO of MERU-VasImmune, Inc., which may develop commercial products based on methodologies presented in this article. S.K., T.K., Y.K., and A.Z. have equity interest in MERU-VasImmune, Inc., and Y.K. is a part-time employee of the company. The remaining authors declare no competing financial interests.
S.K. performed part of this work while at the Blood Research Institute.
Correspondence: Zaverio M. Ruggeri, The Scripps Research Institute, MEM-175, 10550 North Torrey Pines Rd, La Jolla, CA 92037; e-mail: ruggeri@scripps.edu; and Robert R. Montgomery, Blood Research Institute, BloodCenter of Wisconsin, 8733 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: bob.montgomery@bcw.edu.

