

Key points
This is the first prospective, randomized study investigating ECP as a first-line therapy in cGVHD using the 2015 NIH consensus criteria
QoL worsened in patients treated with SoC but remained unchanged in patients treated with SoC+ECP.

Abstract
The investigation of extracorporeal photopheresis (ECP) plus standard of care (SoC) (SoC+ECP) in chronic graft-versus-host disease (cGVHD) within prospective, randomized clinical studies is limited, despite its frequent clinical use. This phase 1/pilot study was the first randomized, prospective study to investigate ECP use as first-line therapy in cGVHD, based on the 2015 National Institutes of Health (NIH) consensus criteria for diagnosis and response assessment. Adult patients with new-onset (≤3 years of hematopoietic stem cell transplantation) moderate or severe cGVHD were randomized 1:1 to 26 weeks of SoC+ECP vs SoC (corticosteroids and cyclosporine A/tacrolimus) between 2011 and 2015. The primary endpoint was overall response rate (ORR), defined as complete or partial response, at week 28 in the intention-to-treat population (ITT). Other outcomes included quality of life (QoL) measures and safety. Sixty patients were randomized; ITT included 53 patients (SoC+ECP: 29; SoC: 24). Week 28 ORR was 74.1% (SoC+ECP) and 60.9% (SoC). Investigator-assessed ORR was 56.0% (SoC+ECP) and 66.7% (SoC). Patients treated with SoC experienced a decline in QoL over the 28-week study period; QoL remained unchanged in SoC+ECP patients. Most frequent treatment-emergent adverse events (TEAEs) in SoC+ECP patients were hypertension (31.0%), cough (20.7%), dyspnea (17.2%), and fatigue (17.2%). Seventeen patients (SoC+ECP: 8; SoC: 9) experienced 35 serious adverse events (SAEs). No TEAEs or SAEs were considered related to the ECP instrument or methoxsalen. The encouraging short-term results of this study could inform the design of subsequent studies. This trial was registered at www.clinicaltrials.gov as #NCT01380535.
Introduction
Chronic graft-versus-host disease (cGVHD) is a complication that occurs in 30% to 40% of patients who have undergone allogeneic hematopoietic stem cell transplantation (HSCT).1,2 The understanding of the pathogenesis of cGVHD continues to evolve and includes donor T-cell alloreactivity and B-cell dysfunction.3 The phenotype is due to inflammation and varying degrees of fibrosis/sclerosis in various organs, such as the oral, esophageal, musculoskeletal, joint, fascial, ocular, lymphohematopoietic systems, and genital tissues.4-6
Due to pleiotropic organ manifestations and varied diagnostic criteria, cGVHD is a complicated disease to diagnose. In 2005, the National Institutes of Health (NIH) introduced consensus criteria for the diagnosis and severity grading of cGVHD in clinical studies, which were further refined in 2015.7 The NIH consensus criteria provide a consistent lexicon and methodology for diagnosing and grading the severity of cGVHD.7
Patients with cGVHD require intensive medical management, which adversely impacts patients’ quality of life (QoL), and long-term morbidity and mortality.8,9 Measurements of QoL highlight the impact of the functional limitations associated with cGVHD on daily activities and typically encompass assessment of overall QoL, physical, emotional, social, and role functioning.10 Reduced QoL is increasingly recognized as an important feature of GVHD and one that requires greater understanding to address the overall impact on patients.11 In observational studies, almost a third of patients, having undergone HSCT, were considered to have poor QoL, and significant differences were typically observed in patients with moderate or severe GVHD.8,12-15 Improvements in QoL have been observed over time in patients following HSCT, but only in those patients without cGVHD.16 There are no large, well-conducted randomized studies investigating the impact of therapeutic intervention on QoL in cGVHD.
The current standard of care (SoC) for cGVHD is corticosteroid prednisone, with a starting dose of 1 mg/kg per day, often in combination with a calcineurin inhibitor.17 However, long-term treatment with corticosteroids can substantially influence patients’ QoL due to the vast array of side effects. Furthermore, high failure rates have been observed in patients with cGVHD receiving first-line treatment with corticosteroids, alone or combined with other immunosuppressants.1,18,19 Consequently, there is an unmet medical need for new first-line treatment approaches for cGVHD.
Extracorporeal photopheresis (ECP) therapy was approved by the Food and Drug Administration (FDA) in cutaneous T-cell lymphoma and has been used for the management of cGVHD since first being described in 1994.20 More recently, recommendations for the use of ECP in GVHD have been published within treatment guidelines, such as those developed by the Stem Cell Transplant Committee.21 Treatment with ECP is an effective second-line therapy in steroid-refractory or steroid-dependent patients with cGVHD.22-24 However, despite the current widespread use of ECP in the treatment of patients with cGVHD, clinical data from randomized studies are limited.22,23 The first prospective, randomized controlled study of ECP as a second-line therapy in steroid-refractory or steroid-dependent cGVHD patients demonstrated that ECP was generally well tolerated and had a steroid-sparing effect over 12 weeks.22 However, ECP treatment as a first-line therapeutic option is yet to be investigated in a randomized controlled trial.1,17
Here, we report data from the randomized, active comparator-controlled, parallel group study of patients with moderate or severe cGVHD receiving first-line treatment with SoC+ECP, vs SoC, with efficacy established using the NIH consensus diagnostic and response criteria.7,25,26 The objective of this hypothesis-generating pilot study was to study the implementation of the NIH Consensus criteria for staging, grading, and response assessment in a multicenter, prospective, randomized study to obtain information and generate better estimates of the primary endpoint, to inform future studies. This study aimed to evaluate the efficacy, safety, and impact on patients’ QoL of SoC+ECP treatment, compared with SoC.
Materials and methods
Patients
Patients were enrolled in Austria, France, Germany, Italy, Hungary, Spain, the United Kingdom, and the United States. Enrollment started in November 2011, and the last patient visit occurred in March 2015.
Key inclusion criteria were new-onset (≤3 years from HSCT) moderate or severe cGVHD as defined by the NIH consensus criteria clinical assessment; the full details are listed in supplemental Table 1. The NIH criteria underwent an interactive refinement from 2005 to 2015.7,27 The study was originally designed to assess response using 2005 criteria. All the variables used in 2015 criteria were captured in the Case Report forms at the time of study initiation. Thus, we report the outcomes using the latest version (2015) of NIH criteria. Key exclusion criteria were intolerance to corticosteroids, previous treatment with prednisone (>2.0 mg/kg per day, 7 days prior to baseline, or >0.5 mg/kg per day 2 weeks prior to screening) or equivalent, or hypersensitivity to methoxsalen; full details are listed in supplemental Table 1.
Written informed consent was obtained from all patients, and the study was conducted in conformity with the Declaration of Helsinki, after receiving institutional review board ethical approval.
Study design
This was a randomized, multicenter, active comparator-controlled, parallel group, pilot study (Figure 1). Patients were randomized 1:1 to receive SoC+ECP or SoC using a central Interactive Voice Response System and were stratified by high risk (platelet count <100 × 109/L at baseline) or low risk.28 SoC consisted of a corticosteroid (prednisone or equivalent) and cyclosporine A or tacrolimus.18 Corticosteroids were started at 1.0 mg/kg per day prednisone or equivalent, not to exceed 2.0 mg/kg. Doses were tapered to 0.5 mg/kg per day by week 8 (±1 week), 0.25 mg/kg per day by week 16 (±1 week), and 0.125 mg/kg per day by week 24 (±1 week); this dose was maintained until week 28. Cyclosporine A and tacrolimus dosing was consistent with local institutional practice to maintain therapeutic blood levels (cyclosporin A 200-300 ng/mL of blood; tacrolimus 4-15 ng/mL of blood), as long as there was no major toxicity.
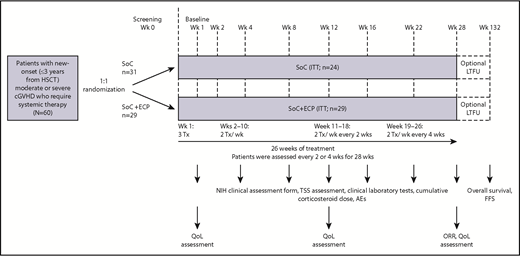
Study design. SoC treatment included 1.0 mg/kg corticosteroids daily (prednisone or equivalent) and CsA or Tac at the usually prescribed dose to maintain the appropriate institutional practice serum level. Corticosteroid doses were tapered to 0.5 mg/kg per day by week (Wk) 8 (±1 week), 0.25 mg/kg per day by week 16 (±1 week), and 0.125 mg/kg per day by week 24 (±1 week); this dose was maintained until week 28. CsA, cyclosporine A; FFS, failure-free survival; LTFU, long-term follow-up; ORR, overall response rate; Tac, tacrolimus; TSS, total skin score; Tx, treatment.
Study design. SoC treatment included 1.0 mg/kg corticosteroids daily (prednisone or equivalent) and CsA or Tac at the usually prescribed dose to maintain the appropriate institutional practice serum level. Corticosteroid doses were tapered to 0.5 mg/kg per day by week (Wk) 8 (±1 week), 0.25 mg/kg per day by week 16 (±1 week), and 0.125 mg/kg per day by week 24 (±1 week); this dose was maintained until week 28. CsA, cyclosporine A; FFS, failure-free survival; LTFU, long-term follow-up; ORR, overall response rate; Tac, tacrolimus; TSS, total skin score; Tx, treatment.
Patients treated with SoC+ECP received ECP therapy according to the following schedule: 3 treatments in the first 7 days, then 2 treatments per week (weeks 2-10), 2 treatments per week every 2 weeks (week 11-18), and 2 treatments per week every 4 weeks (weeks 19-26) (Figure 1). ECP was administered to patients according to the standard instructions provided by Therakos Inc.29 For each treatment, 1500 to 2000 mL of blood was collected, where possible. Methoxsalen was administered at a dosage of 0.017 × treatment volume collected during the plasma/buffy coat collection process. Patients treated with SoC+ECP received all of their treatments with the same Therakos photopheresis system throughout the study (CELLEX or UVAR-XTS). The decision to deliver ECP via a central or peripheral venous catheter was made at the investigators’ discretion and in accordance with individual institutional practice.
Assessments and definitions
For most efficacy analyses, patients were assessed every 2 or 4 weeks for 28 weeks; optional 2-year LTFU was conducted in patients completing the 28-week primary study. The primary efficacy endpoint was ORR at week 28. ORR was defined as clinically assessed complete response (CR) and partial response (PR); the latter required at least 50% improvement on the scale used to measure activity, according to the NIH consensus criteria at week 28.26 Response was assessed by a trained, blinded third-party assessor for skin and mouth domains, as well as the primary physician, to minimize bias in the assessment of patients’ responses. Select secondary efficacy endpoints included TSS change (assessed using the TSS instrument),30 investigator-assessed cGVHD, QoL (assessed at baseline, week 12, and week 28 using the 36-Item Short Form Health Survey [SF-36] and Functional Assessment of Cancer Therapy–Bone Marrow Transplantation [FACT-BMT] surveys),8,31 and cumulative corticosteroid dose at week 28. Validation of the FACT-BMT QoL tool was conducted using the NIH scores for cGVHD (supplemental Table 2).32
Skin assessment by TSS was completed by a trained, blinded third-party assessor and the trained study investigator. Where possible, subjects were evaluated by the same skin assessor for the duration of the study. Blinding was not used in the LTFU portion of the study.
Safety was also evaluated throughout the study. Adverse events (AEs) were categorized by system organ class and preferred term, coded according to the Medical Dictionary for Regulatory Activities (MedDRA), version 16.0. All AEs were reported after provision of informed consent until study completion at week 28 and were monitored throughout the study by an internal medical designee and the external Data Safety and Monitoring Board. The primary endpoint of the LTFU was overall survival in the intention-to-treat (ITT) population at 24 months. Similarly, a secondary endpoint for the LTFU was FFS (lack of GVHD recurrence, new immunosuppressive therapy, relapse, or death from any cause), which was reported for the safety population.
Statistical analysis
This pilot study did not include prespecified hypotheses to test, nor was it powered to do so. The sample size of 60 patients within this pilot study was determined by budget limitations. The ITT population was defined as all randomized patients following the principle of ITT. Subjects were included in the ITT analysis if they had a baseline measurement available corresponding to a posttreatment assessment, according to the treatment to which they were randomized. All efficacy analyses were based on the ITT population. All safety analyses were based on the safety population, defined as all randomized patients who received ≥1 dose of study treatment.
The analysis of the primary endpoint, ORR at week 28, consisted of estimated CR and PR rates with 95% exact binomial confidence intervals (CIs) for both treatment arms. Continuous data were summarized by treatment group using descriptive statistics (mean, median, standard deviation [SD]). Categorical data were summarized by treatment group using frequency tables (frequencies and percentages). Where the data permitted, 95% CI were estimated for all point estimates.
Missing data were not statistically imputed and were treated as failures (no-response); however, last observation carried forward (LOCF) was used to assess the primary endpoint. Patients were included in the efficacy analyses as nonresponders if they dropped out of the study early, could not taper corticosteroid doses according to the study protocol, discontinued due to intolerance to cyclosporine A or tacrolimus, or required additional therapy other than increased corticosteroid dose and permitted medications for cGVHD treatment.
Parameters of QoL were reported at baseline, week 12, and week 28 using the FACT-BMT and SF-36 scores, for patients in each cohort. QoL was then analyzed post hoc through time trend analyses, in order to compare treatment arms using linear mixed regression to account for repeated measurements for each patient.33 Baseline score, time, treatment, and interaction between treatment and time were used as covariates. Spearman correlation was further used to study the correlation of organ-specific score with QoL across the entire patient cohort. Associations between steroid dosing and QoL, measured by FACT-BMT and SF-36, were analyzed by linear mixed models. Post hoc analyses were 1-sided and were performed at the 2.5% level of significance, unless otherwise stated.
Safety assessments were compared between SoC+ECP and SoC treated patients, according to type and frequency of AEs and serious adverse events (SAEs), including treatment-emergent adverse events (TEAEs). Descriptive statistics for reported AEs and SAEs were provided at each visit, by number and percentage of patients reporting the AE.
Results
Patients
Of the 60 patients randomized (SoC+ECP: 29; SoC: 31), 53 (SoC+ECP: 29; SoC: 24) were evaluated for efficacy and comprised the ITT population (Figure 2); 7 patients were excluded from the ITT analyses because these patients did not have the primary efficacy assessment at baseline. Patient disposition during the study is reported in Figure 2. Demographic data were similar between the 2 treatment arms (Table 1).
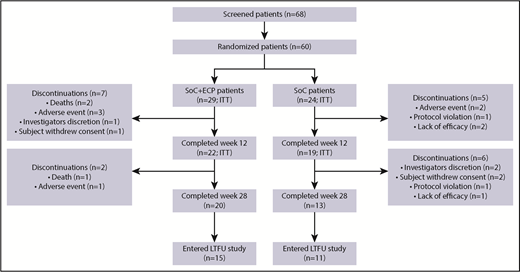
Patient disposition. The proportion of patients who discontinued from the study are shown, along with the proportion of patients who completed the study and those who entered the LTFU study.
Patient disposition. The proportion of patients who discontinued from the study are shown, along with the proportion of patients who completed the study and those who entered the LTFU study.
Patient demographics in ITT patient population
| SoC+ECP (n = 29) | SoC (n = 24) | Overall (n = 53) | |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 21 (72.4) | 14 (58.3) | 35 (66.0) |
| Female | 8 (27.6) | 10 (41.7) | 18 (34.0) |
| Median age (range), y | 51.0 (23-72) | 52.5 (24-68) | 52.0 (23-72) |
| ECOG PS, n (%) | |||
| 0 | 11 (37.9) | 7 (29.2) | 18 (34.0) |
| 1 | 16 (55.2) | 14 (58.3) | 30 (56.6) |
| 2 | 2 (6.9) | 3 (12.5) | 5 (9.4) |
| Primary disease diagnosis, n (%) | |||
| MM | 2 (6.9) | 1 (4.2) | 3 (5.7) |
| MDS | 2 (6.9) | 1 (4.2) | 3 (5.7) |
| NHL | 3 (10.3) | 0 (0.0) | 3 (5.7) |
| ALL | 4 (13.8) | 1 (4.2) | 5 (9.4) |
| AML | 15 (51.7) | 18 (75.0) | 33 (62.3) |
| CLL | 2 (6.9) | 1 (4.2) | 3 (5.7) |
| Other | 1 (3.4) | 2 (8.3) | 3 (5.7) |
| Months from onset of primary disease, mean (SD)* | 27.1 (27.5) | 34.2 (42.3) | 30.3 (34.8) |
| Donor, n (%)† | |||
| HLA-matched relative | 17 (58.6) | 13 (54.2) | 30 (56.6) |
| HLA-mismatched relative (1 Ag) | 1 (3.4) | 0 (0.0) | 1 (1.9) |
| Haploidentical relative | 1 (3.4) | 1 (4.2) | 2 (3.8) |
| Matched unrelated donor | 10 (34.5) | 10 (41.7) | 20 (37.7) |
| Mismatched unrelated donor | 4 (13.8) | 2 (8.3) | 6 (11.3) |
| Stem cell source, n (%) | |||
| Peripheral blood stem cells | 25 (86.2) | 22 (91.7) | 47 (88.7) |
| Bone marrow | 4 (13.8) | 2 (8.3) | 6 (11.3) |
| Months from transplant to first symptoms of cGVHD, mean (SD)* | 6.8 (4.7) | 9.0 (6.0) | 7.8 (5.4) |
| Severity of cGVHD, n (%) | |||
| Moderate | 17 (58.6) | 11 (45.8) | 28 (52.8) |
| Severe | 12 (41.4) | 13 (54.2) | 25 (47.2) |
| Platelet count, n (%) | |||
| <100 × 109/µL (high risk) | 26 (89.7) | 21 (87.5) | 47 (88.7) |
| ≥100 × 109/L (low risk) | 3 (10.3) | 3 (12.5) | 6 (11.3) |
| Organs involved in cGVHD, n (%) | |||
| Skin | 15 (51.7) | 12 (50.0) | 27 (50.9) |
| Liver | 4 (13.8) | 1 (4.2) | 5 (9.4) |
| GI tract | 7 (24.1) | 3 (12.5) | 10 (18.9) |
| Other | 5 (17.2) | 0 (0.0) | 5 (9.4) |
| cGVHD onset type, n (%) | |||
| De novo | 14 (48.3) | 14 (58.3) | 28 (52.8) |
| Progressive | 6 (20.7) | 1 (4.2) | 7 (13.2) |
| Quiescent | 9 (31.0) | 9 (37.5) | 18 (34.0) |
| SoC+ECP (n = 29) | SoC (n = 24) | Overall (n = 53) | |
|---|---|---|---|
| Sex, n (%) | |||
| Male | 21 (72.4) | 14 (58.3) | 35 (66.0) |
| Female | 8 (27.6) | 10 (41.7) | 18 (34.0) |
| Median age (range), y | 51.0 (23-72) | 52.5 (24-68) | 52.0 (23-72) |
| ECOG PS, n (%) | |||
| 0 | 11 (37.9) | 7 (29.2) | 18 (34.0) |
| 1 | 16 (55.2) | 14 (58.3) | 30 (56.6) |
| 2 | 2 (6.9) | 3 (12.5) | 5 (9.4) |
| Primary disease diagnosis, n (%) | |||
| MM | 2 (6.9) | 1 (4.2) | 3 (5.7) |
| MDS | 2 (6.9) | 1 (4.2) | 3 (5.7) |
| NHL | 3 (10.3) | 0 (0.0) | 3 (5.7) |
| ALL | 4 (13.8) | 1 (4.2) | 5 (9.4) |
| AML | 15 (51.7) | 18 (75.0) | 33 (62.3) |
| CLL | 2 (6.9) | 1 (4.2) | 3 (5.7) |
| Other | 1 (3.4) | 2 (8.3) | 3 (5.7) |
| Months from onset of primary disease, mean (SD)* | 27.1 (27.5) | 34.2 (42.3) | 30.3 (34.8) |
| Donor, n (%)† | |||
| HLA-matched relative | 17 (58.6) | 13 (54.2) | 30 (56.6) |
| HLA-mismatched relative (1 Ag) | 1 (3.4) | 0 (0.0) | 1 (1.9) |
| Haploidentical relative | 1 (3.4) | 1 (4.2) | 2 (3.8) |
| Matched unrelated donor | 10 (34.5) | 10 (41.7) | 20 (37.7) |
| Mismatched unrelated donor | 4 (13.8) | 2 (8.3) | 6 (11.3) |
| Stem cell source, n (%) | |||
| Peripheral blood stem cells | 25 (86.2) | 22 (91.7) | 47 (88.7) |
| Bone marrow | 4 (13.8) | 2 (8.3) | 6 (11.3) |
| Months from transplant to first symptoms of cGVHD, mean (SD)* | 6.8 (4.7) | 9.0 (6.0) | 7.8 (5.4) |
| Severity of cGVHD, n (%) | |||
| Moderate | 17 (58.6) | 11 (45.8) | 28 (52.8) |
| Severe | 12 (41.4) | 13 (54.2) | 25 (47.2) |
| Platelet count, n (%) | |||
| <100 × 109/µL (high risk) | 26 (89.7) | 21 (87.5) | 47 (88.7) |
| ≥100 × 109/L (low risk) | 3 (10.3) | 3 (12.5) | 6 (11.3) |
| Organs involved in cGVHD, n (%) | |||
| Skin | 15 (51.7) | 12 (50.0) | 27 (50.9) |
| Liver | 4 (13.8) | 1 (4.2) | 5 (9.4) |
| GI tract | 7 (24.1) | 3 (12.5) | 10 (18.9) |
| Other | 5 (17.2) | 0 (0.0) | 5 (9.4) |
| cGVHD onset type, n (%) | |||
| De novo | 14 (48.3) | 14 (58.3) | 28 (52.8) |
| Progressive | 6 (20.7) | 1 (4.2) | 7 (13.2) |
| Quiescent | 9 (31.0) | 9 (37.5) | 18 (34.0) |
Ag, antigen; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; ECOG, Eastern Cooperative Oncology Group; GI, gastrointestinal; MDS, myelodysplastic syndrome; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; PS, performance status.
Time interval measured in months was calculated as the number of days/30.44.
More than 1 type of donor match was recorded for some patients.
Overall response
In the ITT population, the ORR at 28 weeks was 74.1% (LOCF; 95% CI: 53.7, 88.9) in the SoC+ECP arm and 60.9% (LOCF; 95% CI: 38.5, 80.3) in the SoC arm using the 2015 NIH consensus criteria for cGVHD by blinded assessors (Figure 3A).26 Investigator-assessed ORR was 56.0% (95% CI: 34.9, 75.6) in SoC+ECP patients, and 66.7% (95% CI: 43.0, 85.4) in SoC patients (Figure 3B). There were no statistically significant differences between the SoC+ECP and SoC treatment groups when assessments were blinded or conducted by investigators (Figure 3A-B).
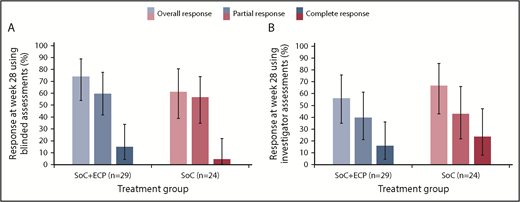
ORR at week 28, according to NIH cGVHD 2015 response criteria using blinded or investigator assessments. LOCF data are reported for the ITT population (n = 53) using blinded assessments (A) and investigator assessments (B). Error bars represent 95% CI calculated using the exact method of binomial distribution. Fisher’s exact test was used to compare the ORRs between patients treated with SoC compared with SoC+ECP; blinded assessments, P = .373; investigator assessments, P = .551.
ORR at week 28, according to NIH cGVHD 2015 response criteria using blinded or investigator assessments. LOCF data are reported for the ITT population (n = 53) using blinded assessments (A) and investigator assessments (B). Error bars represent 95% CI calculated using the exact method of binomial distribution. Fisher’s exact test was used to compare the ORRs between patients treated with SoC compared with SoC+ECP; blinded assessments, P = .373; investigator assessments, P = .551.
In the ITT population, patients with a platelet count of >100 × 109/L at baseline were classified as low risk (47/53, 88.7%), whereas patients with a platelet count of <100 × 109/L (6/53, 11.3%) at baseline were considered to be high risk.28 No significant difference in ORR between SoC+ECP (n = 26; 89.7%) and SoC (n = 21; 87.5%) was observed in the low-risk group; there were insufficient numbers of patients to assess treatment groups separately within the high-risk patients (n = 6).
An overall response at week 28, measured by blinded assessors, was achieved by 75.0% (95% CI: 47.6, 92.7) of patients with moderate cGVHD treated with SoC+ECP and 50.0% (95% CI: 18.7, 81.3) of patients treated with the SoC (Figure 4A). Similarly, an overall response was achieved by 72.7% (95% CI: 39.0, 94.0) of patients with severe cGVHD in the SoC+ECP arm and 69.2% (95% CI: 38.6, 90.9) of patients in the SoC arm (Figure 4C).
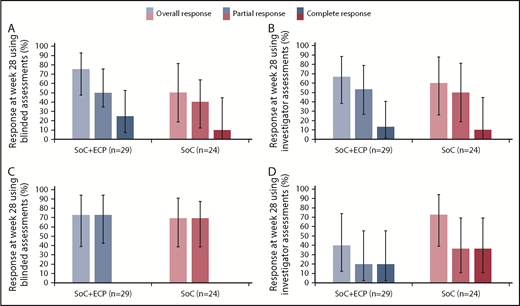
ORR according to NIH cGVHD 2015 response criteria using blinded and investigator assessments at week 28 by severity. LOCF data are reported for the ITT population (n = 53) for patients with moderate cGVHD using blinded (A) and investigator (B) assessments and severe cGVHD using blinded (C) and investigator (D) assessments. Percentages are based on the number of patients treated in the relevant treatment group with no missing responses. Error bars represent 95% CI calculated using the exact method of binomial distribution. Fisher’s exact test was used to compare the ORRs between patients treated with SoC+ECP compared with SoC; blinded assessments in moderate cGVHD patients, P = .234; and severe cGVHD patients, P > .999; investigator assessments in moderate cGVHD patients, P > .999; and severe cGVHD patients, P = .198.
ORR according to NIH cGVHD 2015 response criteria using blinded and investigator assessments at week 28 by severity. LOCF data are reported for the ITT population (n = 53) for patients with moderate cGVHD using blinded (A) and investigator (B) assessments and severe cGVHD using blinded (C) and investigator (D) assessments. Percentages are based on the number of patients treated in the relevant treatment group with no missing responses. Error bars represent 95% CI calculated using the exact method of binomial distribution. Fisher’s exact test was used to compare the ORRs between patients treated with SoC+ECP compared with SoC; blinded assessments in moderate cGVHD patients, P = .234; and severe cGVHD patients, P > .999; investigator assessments in moderate cGVHD patients, P > .999; and severe cGVHD patients, P = .198.
An overall response at week 28, measured by investigators, was achieved by 66.7% (95% CI: 38.4, 88.2) of patients in the SoC+ECP arm and 60.0% (95% CI: 26.2, 87.8) of SoC subjects with moderate cGVHD (Figure 4B). In patients with severe cGVHD, investigator assessments showed that 40.0% (95% CI: 12.2, 73.8) of SoC+ECP patients had an overall response, compared with 72.7% (95% CI: 39.0, 94.0) of SoC patients (Figure 4D).
Changes in TSS
Mean changes (SD) in TSS from baseline to week 28 were similar between treatment arms, by both blinded (SoC+ECP: −0.22 [0.44], SoC: −0.34 [0.53]; P = .549) and investigator assessment (SoC+ECP: −0.27 [0.29], SoC: −0.37 [0.43]; P = .856). In both groups, there was a decline in TSS.
Mean cumulative corticosteroid dose
The mean cumulative corticosteroid dose up to week 28 was similar between treatment arms (4319 mg in the SoC+ECP arm [range: 32-8465 mg]; 4035 mg in the SoC arm [range: 800-7260 mg]).
QoL measurements
For FACT-BMT, no differences were observed in QoL scores over time for patients treated with SoC or SoC+ECP (Figure 5A-B). However, post hoc time trend analyses of the ITT population revealed worsening of QoL in FACT-BMT measures of physical well-being (−0.7326; P = .032), emotional well-being (−0.7151; P = .006), and Functional Assessment of Cancer Therapy–General (−1.6618; P = .018) in the SoC arm. No changes in any QoL domains were observed in SoC+ECP patients over 28 weeks (Figure 5C; supplemental Table 3).
![Figure 5. QoL assessment using FACT-BMT at baseline, week 12, and week 28 in SoC patients, SoC+ECP patients, and time trend analysis for FACT-BMT subscores. Patients completed the FACT-BMT questionnaire for the assessment of QoL at baseline, week 12, and week 28. Median values for SoC patients (A) and SoC+ECP patients (B) are provided. Time trend analyses were performed post hoc. Physical well-being, social/family well-being, and functional well-being were scored from 0 to 28; emotional well-being was scored from 0 to 24, and BMT-specific concerns were scored from 0 to 40. FACT-G Total score (physical well-being + social well-being + functional well-being + emotional well-being [score: 0-108]); FACT-BMT Total score (physical well-being + social well-being + functional well-being + emotional well-being + BMT specific concerns [score: 0-148]); FACT-BMT TOI (physical well-being + functional well-being + BMT-specific concerns [score: 0-96]). (C) Negative estimate denotes decrease of QoL over time. BMT, bone marrow transplantation; SE, standard error; TOI, trial outcome index.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/3/14/10.1182_bloodadvances.2019000145/2/m_advancesadv2019000145f5.png?Expires=1770040930&Signature=lr-jl9I8QKu6m3~u6-22mxlYfl5-1UVx~bbsdWpeCtrOYgALimUBwLnmwnCgj25vtRFZ35UZYprWvObm~URA46zo33Tsb-LEFKSbgiArH4iw3c5zacTLdzAHYyK8iuBNmithtw-ZLxOE-j60pzrsHLnA96xaVHTQYMFNYMdoPSq048nZy4RyMkvacYPTJEDhOruSUcOsouLwhyi~BatX4zGWVVmUIZBfP-C6UhJOUmumidsKnqmY-8w4hz41BATMVyPSp4WUPmt39m61z~syeGafgUVykaa--DTXI7ZAjv0ai9wwKZPM7pyeEMNo8oiKpddqHO0FnrQPfIXjPLO7ng__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
QoL assessment using FACT-BMT at baseline, week 12, and week 28 in SoC patients, SoC+ECP patients, and time trend analysis for FACT-BMT subscores. Patients completed the FACT-BMT questionnaire for the assessment of QoL at baseline, week 12, and week 28. Median values for SoC patients (A) and SoC+ECP patients (B) are provided. Time trend analyses were performed post hoc. Physical well-being, social/family well-being, and functional well-being were scored from 0 to 28; emotional well-being was scored from 0 to 24, and BMT-specific concerns were scored from 0 to 40. FACT-G Total score (physical well-being + social well-being + functional well-being + emotional well-being [score: 0-108]); FACT-BMT Total score (physical well-being + social well-being + functional well-being + emotional well-being + BMT specific concerns [score: 0-148]); FACT-BMT TOI (physical well-being + functional well-being + BMT-specific concerns [score: 0-96]). (C) Negative estimate denotes decrease of QoL over time. BMT, bone marrow transplantation; SE, standard error; TOI, trial outcome index.
QoL assessment using FACT-BMT at baseline, week 12, and week 28 in SoC patients, SoC+ECP patients, and time trend analysis for FACT-BMT subscores. Patients completed the FACT-BMT questionnaire for the assessment of QoL at baseline, week 12, and week 28. Median values for SoC patients (A) and SoC+ECP patients (B) are provided. Time trend analyses were performed post hoc. Physical well-being, social/family well-being, and functional well-being were scored from 0 to 28; emotional well-being was scored from 0 to 24, and BMT-specific concerns were scored from 0 to 40. FACT-G Total score (physical well-being + social well-being + functional well-being + emotional well-being [score: 0-108]); FACT-BMT Total score (physical well-being + social well-being + functional well-being + emotional well-being + BMT specific concerns [score: 0-148]); FACT-BMT TOI (physical well-being + functional well-being + BMT-specific concerns [score: 0-96]). (C) Negative estimate denotes decrease of QoL over time. BMT, bone marrow transplantation; SE, standard error; TOI, trial outcome index.
For SF-36, no differences were observed in QoL scores over time for patients treated with SoC or SoC+ECP (Figure 6A-B). Post hoc time trend analyses showed no significant changes were observed in the SoC+ECP arm, whereas scores for the bodily pain domain significantly worsened by −2.3728 (P = .009) in the SoC arm (ITT population; Figure 6C).
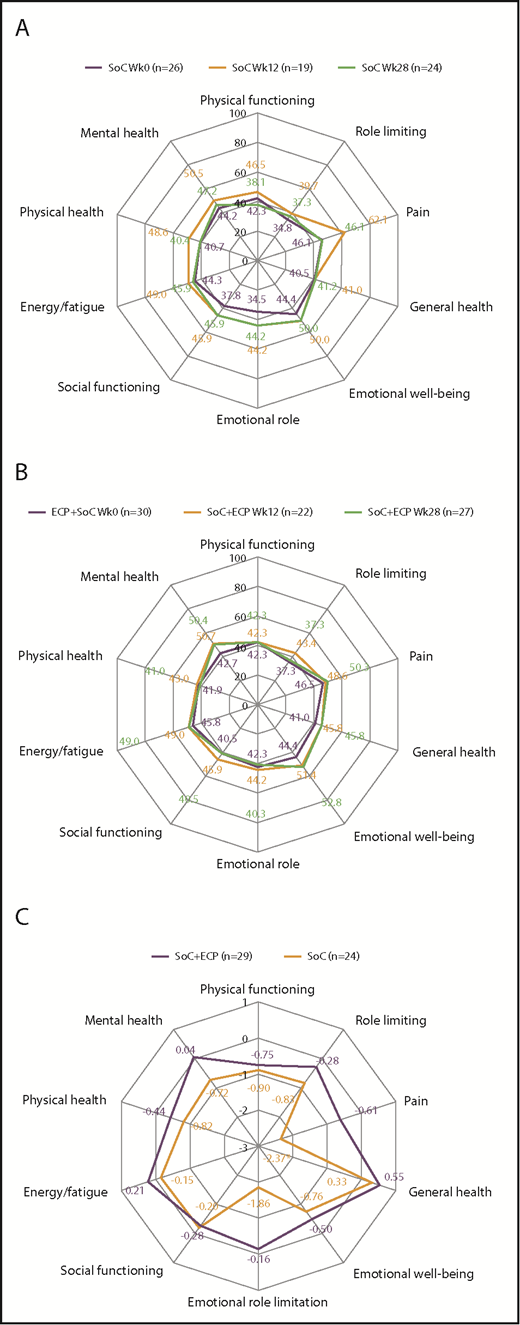
QoL assessment using SF-36 at baseline, week 12, and week 28 in SoC patients, SoC+ECP patients, andtime trend analysis for SF-36 subscores. Patients completed the SF-36 questionnaire for the assessment of QoL at baseline, week 12, and week 28. Median values for SoC patients (A) and SoC+ECP patients (B) are provided. Time trend analyses were performed post hoc. The SF-36 domains of QoL were scored from 0 to 100. (C) Negative estimate denotes decrease of QoL over time. *P < .009.
QoL assessment using SF-36 at baseline, week 12, and week 28 in SoC patients, SoC+ECP patients, andtime trend analysis for SF-36 subscores. Patients completed the SF-36 questionnaire for the assessment of QoL at baseline, week 12, and week 28. Median values for SoC patients (A) and SoC+ECP patients (B) are provided. Time trend analyses were performed post hoc. The SF-36 domains of QoL were scored from 0 to 100. (C) Negative estimate denotes decrease of QoL over time. *P < .009.
Correlation analyses of QoL with steroid dose were conducted, and despite a reduction of FACT-BMT, total score between week 12 and week 24 in SoC patients, changes in steroid dose were not associated with change in QoL (P > .05 for all parameters of QoL), as determined by a linear mixed model. In the SoC+ECP arm, changes in steroid dose were associated with beneficial changes in physical, emotional, and functional well-being, FACT-G, and FACT-BMT (total and TOI) (supplemental Table 4). Similarly, for SF-36, changes in steroid dose were associated with beneficial changes in general health, emotional well-being, social functioning, and mental health in patients in the SoC+ECP arm; however, no associations were observed in SoC patients (supplemental Table 5).
FFS: 2-year LTFU patients only (secondary endpoint)
Among the 32 patients in the safety population who underwent LTFU, the median duration of FFS was 12.5 months (95% CI: 4.8, 22.4) in the SoC+ECP arm, compared with 7.8 months (95% CI: 1.8, 19.5) in the SoC arm (P = .611; supplemental Figure 1). A total of 13/32 patients failed due to the addition of a new immunosuppressive therapy (ECP+SoC: 7 patients; SoC: 6 patients); 3/32 patients failed due to relapse of their original malignancy (ECP+SoC: 1 patient; SoC: 2 patients), and 4/32 patients failed due to recurrence of cGVHD (ECP+SoC: 3 patients; SoC: 1 patient).
Safety
All 60 patients were evaluated for safety. Overall, 96.6% of SoC+ECP–treated patients and 90.3% of SoC-treated patients experienced TEAEs (Table 2). The number of events was lower in the SoC+ECP arm (n = 223) than in the SoC arm (n = 316). However, because of the small number of subjects in each treatment arm, these data should be interpreted with caution. The most frequently reported TEAEs in the SoC+ECP arm were hypertension (31.0%), cough (20.7%), dyspnea (17.2%), and fatigue (17.2%), whereas the most common event in the SoC arm was back pain (16.1%) (Table 2). Deep vein thrombosis was not reported by patients treated with SoC+ECP, but was reported by 2 (6.5%) SoC patients. No cases of pulmonary embolism were reported during this study.
Summary of AEs in either treatment arm
| Event, n (%) [no. of reported AEs], unless otherwise stated | SoC+ECP (n = 29) | SoC (n = 31) | Total (n = 60) |
|---|---|---|---|
| Any AE | 28 (96.6) [223] | 28 (90.3) [316] | 56 (93.3) [539] |
| Any TEAE | 28 (96.6) [223] | 28 (90.3) [316] | 56 (93.3) [539] |
| TEAEs occurring in ≥10% of patients,*n (%) | |||
| Hypertension | 9 (31.0) | 4 (12.9) | 13 (21.7) |
| Cough | 6 (20.7) | 1 (3.2) | 7 (11.7) |
| Dyspnea | 5 (17.2) | 2 (6.5) | 7 (11.7) |
| Fatigue | 5 (17.2) | 1 (3.2) | 6 (10.0) |
| Hyperglycemia | 4 (13.8) | 4 (12.9) | 8 (13.3) |
| Increase of γ-glutamyltransferase | 4 (13.8) | 3 (9.7) | 7 (11.7) |
| Dizziness | 4 (13.8) | 2 (6.5) | 6 (10.0) |
| Decrease of platelet count | 3 (10.3) | 2 (6.5) | 5 (8.3) |
| Increase of blood cholesterol | 3 (10.3) | 1 (3.2) | 4 (6.7) |
| Hypertriglyceridemia | 3 (10.3) | 1 (3.2) | 4 (6.7) |
| Hypokalemia | 3 (10.3) | 1 (3.2) | 4 (6.7) |
| Dry mouth | 3 (10.3) | 0 | 3 (5.0) |
| Back pain | 2 (6.9) | 5 (16.1) | 7 (11.7) |
| Muscle spasms | 2 (6.9) | 4 (12.9) | 6 (10.0) |
| Peripheral edema | 2 (6.9) | 4 (12.9) | 6 (10.0) |
| Thrombocytopenia | 2 (6.9) | 4 (12.9) | 6 (10.0) |
| Diarrhea | 1 (3.4) | 4 (12.9) | 5 (8.3) |
| Pyrexia | 0 | 4 (12.9) | 4 (6.7) |
| Any treatment-related AE | 21 (72.4) [54] | 20 (64.5) [71] | 41 (68.3) [125] |
| Severe AE | 10 (34.5) [16] | 11 (35.5) [26] | 21 (35.0) [42] |
| Serious AE | 8 (27.6) [18] | 9 (29.0) [17] | 17 (28.3) [35] |
| Treatment-related SAE, n (%) | 6 (20.7) | 7 (22.6) | 13 (21.7) |
| Patients with any treatment-related SAEs leading to withdrawal,† n (%) | 6 (20.7) | 6 (19.4) | 12 (20.0) |
| Infections and infestations | 5 (17.2) | 2 (6.5) | 7 (11.7) |
| Respiratory, thoracic, and mediastinal disorders | 3 (10.3) | 0 | 3 (5.0) |
| Metabolism and nutrition disorders | 1 (3.4) | 1 (3.2) | 2 (3.3) |
| Musculoskeletal and connective tissue disorders | 0 | 2 (6.5) | 2 (3.3) |
| Psychiatric disorders | 0 | 1 (3.2) | 1 (1.7) |
| Life-threatening AE | 6 (20.7) [8] | 2 (6.5) [2] | 8 (13.3) [10] |
| Death | 4 (13.8) [4] | 0 | 4 (6.7) [4] |
| Event, n (%) [no. of reported AEs], unless otherwise stated | SoC+ECP (n = 29) | SoC (n = 31) | Total (n = 60) |
|---|---|---|---|
| Any AE | 28 (96.6) [223] | 28 (90.3) [316] | 56 (93.3) [539] |
| Any TEAE | 28 (96.6) [223] | 28 (90.3) [316] | 56 (93.3) [539] |
| TEAEs occurring in ≥10% of patients,*n (%) | |||
| Hypertension | 9 (31.0) | 4 (12.9) | 13 (21.7) |
| Cough | 6 (20.7) | 1 (3.2) | 7 (11.7) |
| Dyspnea | 5 (17.2) | 2 (6.5) | 7 (11.7) |
| Fatigue | 5 (17.2) | 1 (3.2) | 6 (10.0) |
| Hyperglycemia | 4 (13.8) | 4 (12.9) | 8 (13.3) |
| Increase of γ-glutamyltransferase | 4 (13.8) | 3 (9.7) | 7 (11.7) |
| Dizziness | 4 (13.8) | 2 (6.5) | 6 (10.0) |
| Decrease of platelet count | 3 (10.3) | 2 (6.5) | 5 (8.3) |
| Increase of blood cholesterol | 3 (10.3) | 1 (3.2) | 4 (6.7) |
| Hypertriglyceridemia | 3 (10.3) | 1 (3.2) | 4 (6.7) |
| Hypokalemia | 3 (10.3) | 1 (3.2) | 4 (6.7) |
| Dry mouth | 3 (10.3) | 0 | 3 (5.0) |
| Back pain | 2 (6.9) | 5 (16.1) | 7 (11.7) |
| Muscle spasms | 2 (6.9) | 4 (12.9) | 6 (10.0) |
| Peripheral edema | 2 (6.9) | 4 (12.9) | 6 (10.0) |
| Thrombocytopenia | 2 (6.9) | 4 (12.9) | 6 (10.0) |
| Diarrhea | 1 (3.4) | 4 (12.9) | 5 (8.3) |
| Pyrexia | 0 | 4 (12.9) | 4 (6.7) |
| Any treatment-related AE | 21 (72.4) [54] | 20 (64.5) [71] | 41 (68.3) [125] |
| Severe AE | 10 (34.5) [16] | 11 (35.5) [26] | 21 (35.0) [42] |
| Serious AE | 8 (27.6) [18] | 9 (29.0) [17] | 17 (28.3) [35] |
| Treatment-related SAE, n (%) | 6 (20.7) | 7 (22.6) | 13 (21.7) |
| Patients with any treatment-related SAEs leading to withdrawal,† n (%) | 6 (20.7) | 6 (19.4) | 12 (20.0) |
| Infections and infestations | 5 (17.2) | 2 (6.5) | 7 (11.7) |
| Respiratory, thoracic, and mediastinal disorders | 3 (10.3) | 0 | 3 (5.0) |
| Metabolism and nutrition disorders | 1 (3.4) | 1 (3.2) | 2 (3.3) |
| Musculoskeletal and connective tissue disorders | 0 | 2 (6.5) | 2 (3.3) |
| Psychiatric disorders | 0 | 1 (3.2) | 1 (1.7) |
| Life-threatening AE | 6 (20.7) [8] | 2 (6.5) [2] | 8 (13.3) [10] |
| Death | 4 (13.8) [4] | 0 | 4 (6.7) [4] |
Data are reported for the safety population. AEs and SAEs were categorized using MedDRA version 16.0. TEAEs were defined as AEs that started after study drug administration on day 1, or reemerged or worsened during treatment. SAEs were defined as any untoward medical occurrence that, at any dose, is life-threatening, requires or prolongs in-patient hospitalization, or results in a persistent or significant disability, congenital anomaly or birth defect, an important medical event, or death.
TEAEs reported by MedDRA Preferred Term, where ≥10% of patients in either the SoC+ECP or the SoC group reported the TEAE (indented values).
SAEs leading to withdrawal are reported by MedDRA System Organ Class (indented values).
Overall, 17 patients experienced 35 SAEs. In the SoC+ECP arm, 18 events occurred in 8 (27.6%) patients, whereas in the SoC arm, 17 events were observed in 9 (29.0%) patients. SAEs in 13 patients (21.7%) were considered related to study treatment; however, no events were considered related to the ECP instrument or methoxsalen. Six patients from each arm (SoC+ECP: 20.7%; SoC: 19.4%) were withdrawn from the study due to SAEs (Table 2).
A total of 20 (37.7%) patients discontinued the primary study (SoC+ECP: 9 [31.0%] and SoC: 11 [45.8%]). In the SoC+ECP arm, 4 (13.8%) patients discontinued early due to AEs, and 3 (10.3%) patients discontinued due to death, whereas in the SoC arm, 2 (8.3%) patients discontinued early due to AEs and 0 patients discontinued due to death.
Four deaths occurred in patients randomized to the SoC+ECP arm during the primary study; two of these patients had moderate cGVHD and two had severe cGVHD. The causes of death were 1 case each of acute heart failure due to Escherichia coli sepsis and acute respiratory distress syndrome, and 2 cases of sepsis. Three of these deaths occurred during active treatment, and 1 patient died after withdrawing from the study. Three of these deaths were considered related to treatment with corticosteroid and/or calcineurin inhibitor therapy, and 1 death was not considered related to any study treatment. None of the deaths were considered to be related to the instrument, ECP therapy, or methoxsalen.
Discussion
The present results suggest that ECP with methoxsalen is a well-tolerated first-line treatment of cGVHD in patients who have undergone HSCT. To our knowledge, the impact of cGVHD on patients’ QoL has not been addressed in a prospective, interventional study comparing ECP+SoC treatment with SoC alone; therefore, this study adds to the currently limited cGVHD QoL literature.8,15
The results of this study are important in planning future studies and for clinical study design approaches because we provide additional information to base future point and variability estimates. In August 2017, ibrutinib was approved in the United States for adult patients with cGVHD who have failed to respond to ≥1 systemic therapy, based on the results of a single-arm, phase 2 study.34,35 Similar to the present study assessing ECP, the primary efficacy response of cGVHD response was assessed using the NIH consensus response criteria.7,26 The FDA has not approved any drugs or devices for first-line therapy of moderate to severe cGVHD. In the present study, the SoC+ECP arm showed an encouraging ORR of 74.1% at week 28, using an ITT approach, compared with 60.9% in the SoC arm. Future superiority studies reporting ORR will require larger sample sizes to achieve statistical significance. An ongoing phase 3 study comparing ibrutinib with corticosteroids (#NCT02959944) for first-line therapy of moderate to severe cGVHD is currently underway. If the SoC ORR from the present study is replicated, then it will be necessary for the ibrutinib ORR to be much higher than the 74.1% achieved in the current study, for a positive result in the first-line setting.
Univariate and multivariate analyses from a prospective cohort study showed that treatment responses were more frequent in cGVHD patients who had a platelet count ≥100 × 109/L (low risk).36 In the present study, >80% of patients had a platelet count <100 × 109/L (high risk), suggesting that response to treatment was not influenced by platelet count; however, statistical analyses were not conducted to determine whether platelet count influenced any of the reported outcomes. Future studies, with larger sample sizes, would ensure a more uniform distribution of platelet counts in each treatment arm. The SoC+ECP group included more patients with progressive onset cGVHD than the SoC group (20.7% vs 4.2%), suggesting that overall the SoC+ECP group included more prognostically inferior patients at baseline.
The few prospective studies investigating ECP in steroid-refractory or steroid-dependent cGVHD patients have demonstrated significant reductions in corticosteroid doses and progressive improvements in cutaneous responses.23,37,38 The first prospective, randomized controlled study of ECP in cGVHD suggested that ECP was well tolerated and had a steroid-sparing effect; however, this study was limited by the fact that a high proportion of patients in the control arm crossed over to ECP treatment.22 However, it is unlikely that steroid sparing would be considered a clinical study endpoint, because the decision making and practice of tapering steroid doses is variable among physicians. Similarly, FFS, although a meaningful endpoint, is subject to variation in the timing when physicians add the next line of therapy to patients with nonresponding or progressive cGVHD. The present study was not designed to assess the difference in FFS between the 2 arms; however, the median duration of FFS in the SoC+ECP arm (12.5 months) was encouraging when compared with the SoC (7.8 months) alone, despite the small sample size (n = 32; SoC+ECP patients: 39%; SoC patients: 36%).
A prospective, multicenter, observational study showed that ∼15% of cGVHD patients had FFS and CR or PR after 1 year of first-line treatment, and 45% to 56% of patients received a secondary systemic treatment prior to the 1-year overall response assessment.1 These findings suggest that cGVHD was inadequately controlled with first-line treatment and that in any first-line study with steroids in the comparator arm, the response in the experimental arm would have to be substantially higher to demonstrate a statistical difference. The data from the present study cannot, therefore, be compared with the existing literature and highlight the need for further prospective studies investigating FFS, CR, and PR in patients with cGVHD.
A key concern of transplant survivors is QoL, and thus, studies reporting the impact of treatment on QoL are of paramount importance. The impact of corticosteroids and tapering regimens on QoL has not been studied extensively in prospective clinical trials. QoL has not been used as a sole primary endpoint or composite primary endpoint in clinical studies of cGVHD due to lack of validated QoL tools. We used standard QoL measures in the present study to determine whether these methods are sensitive enough to detect QoL differences in patients with cGVHD. Patient-reported QoL, in patients with NIH-defined moderate or severe cGVHD, showed that multiple domains, including physical functioning, were significantly compromised when assessed by SF-36 and FACT-BMT; these observations were in line with impairments observed in other autoimmune disorders.8 The lack of impairment of QoL observed in the SoC+ECP arm in the present study is encouraging when compared with patients in the SoC arm who experienced worsening of QoL between week 12 and week 24; however, this study was not powered to detect changes in QoL between the 2 arms, and so these results should be interpreted with caution. In addition, in the SoC+ECP arm, changes in steroid dose were associated with changes in physical, emotional, and functional well-being, FACT-G, and FACT-BMT (total and TOI). Considering the extensive array of side effects associated with high doses of corticosteroids, these findings are of importance to patients; however, the small patient number in each arm in our study is a limitation to these findings.
The FDA had recently issued a concern regarding venous thromboembolism in patients undergoing ECP.39 In this study, no new safety events were identified for treatment with SoC+ECP, and in particular, no cases of venous thromboembolism were reported, whereas 2 cases of deep vein thrombosis were reported in a patient treated with SoC. The observed safety profile was in line with that reported in previous publications for ECP.22,40 None of the deaths reported were considered to be related to the instrument, ECP treatment, or methoxsalen.
This pilot study fulfilled the aim of informing the design of future studies; however, as such, this study was limited by the small sample size, which meant that the study was not statistically powered to detect differences. The follow-up of patients in the primary phase of this pilot trial was only 28 weeks, and thus, a longer follow-up period would be needed to demonstrate the long-term sustainability of results in both treatment arms. The inclusion of patient-reported QoL measurements may result in biased results as the patients were not blinded to their treatment allocation. The strengths of this study were that it was a randomized controlled trial, which included the use of NIH grading for diagnosis to recruit patients, and the NIH response assessment criteria.7,26 The use of trained, blinded assessors to perform assessments was another major strength of this pilot study, as this removed bias from the results; however, it was not possible to blind the patients or their treating physician from the treatment regimen.
In conclusion, treatment with SoC+ECP did not identify any new safety events in this study, and a trend for improvement for QoL was observed in the small SoC+ECP patient population. Treatment with SoC+ECP showed an encouraging ORR at week 28; however, the ORR for SoC patients was also high, highlighting the need for further studies with greater power. This study lays the foundation to design future clinical studies for first-line therapy of NIH-diagnosed moderate to severe cGVHD. The observed ORR using an ITT allows the scientific community to plan appropriately powered future clinical studies and provides a benchmark for ORR in SoC-treated patients.
Results from analyses of additional secondary efficacy endpoints from the main study and LTFU are available at https://www.clinicaltrials.gov/ct2/show/NCT01380535. Researchers whose proposed use of the data has been approved by an independent review committee will be given access to anonymized data that underlie the results reported in this article.
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank the patients and their caregivers in addition to the investigators and their teams who contributed to this study. The authors acknowledge Gregory Tardie and Michael Blasi, both of Mallinckrodt Pharmaceuticals, for publication coordination. The authors also extend thanks to Sarah Jayne Clements (Costello Medical, Cambridge, United Kingdom) for medical writing and editorial assistance in the preparation of this manuscript, which was conducted in accordance with Good Publication Practices and funded by Therakos, Inc, a Mallinckrodt Pharmaceuticals Company.
This study was funded by Therakos, Inc, a Mallinckrodt Pharmaceuticals Company.
The sponsor participated in the review and decision to publish this manuscript.
Authorship
Contribution: M.J., H.T.G., C.S., G.S., F.A.A., J.T., M.L.D., Á.B., H.C., S.-C.C., T.C., H.B., and G.M. provided substantial contributions to study conception and design; provided substantial contributions to acquisitions of data; provided substantial contributions to analysis and interpretation of data; drafted the article or critically revised it for important intellectual content; and provided final approval of the article to be published.
Conflict-of-interest disclosure: M.J. receives research support from Janssen and Mallinckrodt Pharmaceuticals and serves as Consultant for Incyte. H.T.G. receives honoraria from Amgen, Celgene, Novartis, and Roche and serves on the Speaker’s bureau for Therakos. C.S. receives honoraria from Amgen, Bristol-Myers Squibb, Celgene, Janssen, Novartis, and Sanofi. G.S. receives honoraria and lecture fees from Therakos, Inc, a Mallinckrodt Pharmaceuticals Company. F.A.A. receives honoraria and research funding from Therakos, Inc, a Mallinckrodt Pharmaceuticals Company. T.C. and H.B. are employees of Mallinckrodt Pharmaceuticals. G.M. is a former employee of Mallinckrodt Pharmaceuticals. The remaining authors declare no competing financial interests.
Correspondence: Madan Jagasia, Division of Hematology/Oncology, Vanderbilt-Ingram Cancer Center, 3927 The Vanderbilt Clinic, Nashville, TN 37232; e-mail: madan.jagasia@vanderbilt.edu.

