

Key Points
In vivo CRISPR screens in AML define key interactors of the microenvironment, including integrins, immune modulators, and glycosylation.
Eight in vivo–specific hits are recurrently associated with adverse prognosis: BTBD6, FERMT3, ILK, SLC19A1, TAP2, TLN1, TPST2, and TRMT12.

Abstract
Genome-wide CRISPR screens have been extremely useful in identifying therapeutic targets in diverse cancers by defining genes that are essential for malignant growth. However, most CRISPR screens were performed in vitro and thus cannot identify genes that are essential for interactions with the microenvironment in vivo. Here, we report genome-wide CRISPR screens in 2 in vivo murine models of acute myeloid leukemia (AML) driven by the KMT2A/MLLT3 fusion or by the constitutive coexpression of Hoxa9 and Meis1. Secondary validation using a focused library identified 72 genes specifically essential for leukemic growth in vivo, including components of the major histocompatibility complex class I complex, Cd47, complement receptor Cr1l, and the β-4-galactosylation pathway. Importantly, several of these in vivo–specific hits have a prognostic effect or are inferred to be master regulators of protein activity in human AML cases. For instance, we identified Fermt3, a master regulator of integrin signaling, as having in vivo–specific dependency with high prognostic relevance. Overall, we show an experimental and computational pipeline for genome-wide functional screens in vivo in AML and provide a genome-wide resource of essential drivers of leukemic growth in vivo.
Introduction
Acute myeloid leukemia (AML) is a highly lethal hematologic malignancy; ∼70% of patients with AML will ultimately die of the disease.1 Systematic DNA sequencing of AML patient samples has delineated a series of mutated genetic drivers implicated in AML pathogenesis2,3 that collaborate to promote self-renewal and proliferation of leukemic stem cells (LSCs).4 Mapping the mutational landscape in AML has highlighted unique genetic vulnerabilities that have been exploited for targeted antileukemic therapies, such as the inhibitors of mutated proteins FMS-like tyrosine kinase 3 and isocitrate dehydrogenase 1 or 2.5,6 However, only a limited fraction of patients with AML are eligible for these targeted therapies, highlighting the unmet clinical need for identifying additional “druggable” AML drivers.
Targeting essential interactions between leukemic cells and their microenvironment represents a promising avenue for treating AML.7,8 For example, characterization of murine experimental models of AML has highlighted the critical role of the bone marrow microenvironment in mediating the survival of leukemic cells in vivo9 through metabolic rewiring,10-12 immune evasion,13-15 and suppression of normal hematopoiesis.16,17 However, our understanding of the “leukemic interactome in vivo” remains partial, as most experimental models are limited in the numbers of genes that can be tested simultaneously. In this regard, the use of pooled CRISPR/Cas9 screening technology in vivo is an appealing approach because in vitro pooled screens have been very successful in identifying key players in leukemogenesis,18 such as regulators of messenger RNA modifications,19,20 transcription,21,22 and metabolism.23-25 A major technical limitation of in vivo pooled screens is the requirement for engraftment of a large number of cells to minimize false-positive results.26 This difficulty has been recently overcome in AML by limiting the size of the small guide RNA (sgRNA) libraries transplanted in vivo20,27,28 ; however, a systematic genome-wide screen in vivo has yet to be completed in AML.
The current study reports a series of genome-wide CRISPR screens completed both in vivo and in vitro in 2 AML mouse models (KMT2A/MLLT3 [also known as MLL/AF9] and HOXA9/MEIS1). Using this pipeline, we identified in vivo–specific essential genes and validated several factors involved in microenvironmental interactions with AML cells. Using an integrative bioinformatics approach, we identified essential genetic regulators of leukemic stemness and prognosis in human AML. Collectively, we generated a genome-wide map of essentiality for AML in vivo, which provides a systematic resource for identifying prognostic markers and therapeutically relevant targets.
Materials and methods
Murine models
Experiments were approved by the Institutional Animal Care and Use Committee of the Massachusetts General Hospital. B6;129-Gt(ROSA)26Sortm1.1(CAG-cas9*,-EGFP)Fezh (Cas9-GFP) mice were a generous gift from the laboratory of F. Zhang.29
Generation of murine leukemic cell lines
The KMT2A/MLLT3 (MLL/AF9) and HOXA9/MEIS1 retroviral leukemia models have been described previously.30,31 The MLL/AF9 Cas9-GFP (“MA”) line was established by sorting multipotent progenitors (Lineage–c-KithighSca-1–) from a male Cas9-GFP+/− donor with a MSCV-MLL/AF9-IRES-GFP construct.30 The HOXA9/MEIS1 Cas9-GFP (“HM”) line was established by sorting Lineage– progenitors from a male Cas9-GFP+/− donor and transducing with a MSCV-HoxA9-IRES-Meis1 construct.31 Respective progenitors were sorted in transduction medium consisting of RPMI 1640 (Lonza) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 IU/mL penicillin, 100 mg/mL streptomycin (Gibco), 10 ng/mL recombinant mouse stem cell factor, 6 ng/mL recombinant mouse interleukin 3 (rmIL-3), 5 ng/mL rmIL-6 (all from PeproTech), and incubated overnight. The following day, the progenitors were transferred to 6-well plates coated in RetroNectin (Lonza) in a volume of 1 mL, combined with 1 mL of fresh retroviral supernatant packaged in 293FT cells (Invitrogen) with pCL-Eco32 (#12371; Addgene) and 8 µg/mL final concentration of polybrene, and then spinoculation (1000g, 90 minutes, 22°C). Immediately after the transduction, 4 mL of fresh transduction medium was added to each well to dilute the polybrene; 24 hours’ posttransduction, cells were washed by centrifugation (500g, 5 minutes, 4°C) and then resuspended in fresh transduction medium. MA and HM cell lines with no selectable marker emerged after 3 to 4 weeks of continuous passage in culture. This was in contrast to uninfected cells maintained in the same conditions that differentiated and ceased proliferating over this same period.33 MA and HM cell lines were maintained in leukemic culture medium consisting of RPMI 1640 supplemented with 10% FBS, penicillin/streptomycin, 6 ng/mL rmIL-3, and ∼100 ng/mL stem cell factor generated from a Chinese hamster ovary cell line, as previously described.33
Syngeneic leukemia experiments
All leukemia transplantations were performed by injecting cells IV in a volume of 300 µL of phosphate-buffered saline (PBS) in recipient C57Bl/6J male mice (aged 8-10 weeks) that were sublethally irradiated (4.5 Gy) the day prior using a Cesium-137 source. Blood was collected through tail vein sampling, and complete blood counts were performed on an Element HT5 analyzer (Heska Corporation). Hematopoietic cell isolation was done as follows: after euthanasia by asphyxiation with carbon dioxide, vertebrae and the long bones of the arms and legs were dissected and crushed into ice-cold PBS containing 2% FBS. The bone marrow mononuclear cells were isolated over a Ficoll-Paque PLUS density gradient and counted by using trypan blue and the Cellometer instrument (Nexcelom Bioscience LLC).
In vivo CRISPR screening
To generate the cell lines used in the screen, 5 million MA or HM cells were injected into sublethally irradiated recipients. After development of clinical signs of leukemia, bone marrow cells were harvested as described earlier and replated in vitro. This procedure was repeated an additional time for the HM line. Explanted AML cells were expanded in vitro, then cryopreserved in several aliquots in liquid nitrogen. Ex vivo–expanded MA or HM cells were transduced with the GeCKO (Genome-Scale CRISPR Knock-Out) v2 genome-wide lentiviral libraries comprising 130 209 targeting sequences and 1000 sequences with no homology to the mouse genome as control.34 To diminish the number of sgRNAs per condition, each cell line was transduced separately with each half of the library, named pools A and B. Fresh unconcentrated lentiviral supernatant was generated in 293FT cells (Thermo Fisher Scientific). For each experiment, cells were transduced by aliquoting 24 million cells into four 6-well plates at a ratio of 106 cells/1 mL of media per well, adding 1 mL of lentiviral supernatant and polybrene (8 µg/mL), and then spinoculation (1000g, 90 minutes, 22°C).
Immediately after the transduction, 4 mL of fresh media was added to each well to dilute the polybrene. Twenty-four hours’ posttransduction, cells were washed by centrifugation (500g, 5 minutes, 4°C) then resuspended in 100 mL of fresh media and transferred to T175 flasks. Forty-eight hours’ posttransduction, cells were selected with puromycin 10 µg/mL (Gibco) for 5 days. Cells were passaged to maintain a concentration between 5 × 105 and 1.5 × 106. Seven days after transduction, for each half-library, 4 × 107 MA or HM cells were transplanted per mouse in 20 sublethally irradiated mice (2 retro-orbital injections of 2 × 107 cells, with an interval of 12 hours). Thus, for each pool of sgRNAs, 8 × 108 cells in total were transplanted in vivo. At the same moment, an aliquot of cells was preserved for sequencing, and 2 × 108 cells were maintained in vitro for the same duration as the in vivo arm. After 14 days of in vivo growth, the mice were euthanized, and bone marrow cells were harvested, as described earlier. Cells from each time point were cryopreserved in 90% FBS and 10% dimethyl sulfoxide in aliquots of ∼40 million cells.
sgRNA library sequencing
Illumina-compatible sequencing libraries were generated by using a 1-step polymerase chain reaction (PCR) protocol developed by the Genetic Perturbation Platform of the Broad Institute (available at https://portals.broadinstitute.org/gpp/public/dir/download?dirpath=protocols/production&filename=sgRNA_shRNA_ORF_PCR_for_sequencing_20200619_public.zip). P5 primer mix NEON and P7 indexed primers KERMIT were used. The proviral sgRNAs were amplified at the following time points: (1) seven days in vitro after transduction; (2) an additional 14 days in vitro after time point #1; and (3) 14 days in vivo after time point #1. From each condition, genomic DNA was extracted by using DNAQuik reagents (REPROCELL), and 40 µg of genomic DNA was amplified. Then, 1 ng of plasmid libraries “Gecko A” and “Gecko B” was similarly amplified to provide baseline representation of the sgRNAs within the libraries. PCR products were migrated on an agarose gel, and the expected bands at ∼350 bp were extracted and pooled. Sequencing was performed on a HiSeq 2000 flow cell (Illumina).
Analysis of genome-wide CRISPR dropout screen
We developed a customized analytical workflow to identify essential genes for genome-wide CRISPR screens. For each condition (plasmid, pretransplant, in vitro, and in vivo), read counts from all biological replicates were pooled, followed by global median normalization. Conditions of interest were then compared, and log2 fold changes (log2FC) were obtained for each sgRNA. To assess the biological effect of each gene, the Wilcoxon rank sum test was used to summarize the collective changes of associated sgRNAs, and the result was represented as a z score, which is normally distributed. A positive z score indicates enrichment, and a negative z score indicates depletion. To determine statistical significance, a permutation P value was calculated by comparing observed z score vs a background distribution generated by 1000 control sgRNAs.
Validation of CRISPR screen using a focused library
The 1340 top-scoring in vivo and in vitro genes from the genome-wide CRISPR screen were selected for validation, with filtering for druggable candidates35 and against genes likely to represent essential biological processes.36 A validation library targeting each gene with 10 sgRNAs and containing 1000 nontargeting controls was designed by using the prediction tool of the Broad Institute Genetic Perturbation Platform (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) and cloned into the lentiGuide-Puro vector.34 The in vivo and in vitro validation screen was repeated with the same protocol as for the aforementioned genome-wide screen. From the sgRNA count matrices, essential genes were identified by MAGeCK using control sgRNAs for normalization with a false discovery rate (FDR) of 1%.37
Pathway enrichment analysis
Canonical pathways and hallmark gene sets defined in the Molecular Signatures Database were used for pathway enrichment. P values were calculated by using the hypergeometric test and adjusted for multiple testing by using the Benjamini-Hochberg procedure to control the FDR. For identification of genetic hits linked to the immune system, we downloaded GO:0002376 (http://www.informatics.jax.org/go/term/GO:0002376).
RCA-I lectin staining
Cells were washed once with PBS containing 2% FBS and diluted to 1 × 107 cells/mL. Then, 100 µL of cell suspension was stained with RCA-I (rhodamine conjugated Ricinus communis agglutinin I; 1:1000 dilution; catalog no. RL-1082; Vector Laboratories) for 1 hour at room temperature. As control, cells were incubated with trypsin for 15 minutes at room temperature before staining. Cells were washed with 1x PBS containing 5% FBS and analyzed on a BD Fortessa instrument.
Western blotting
Cells were washed with cold PBS, and whole-cell lysates were collected by using freshly prepared lithium dodecyl sulfate sample buffer containing proteinase and phosphatase inhibitor. Cell lysates were sonicated at 30% for 5 seconds for 3 rounds. Protein concentration was measured by NanoDrop (A280; Thermo Fisher Scientific). Then, 1,4 µL of β-mercaptoethanol (catalog no. M3148; MilliporeSigma) and 4x Laemmli Sample buffer (catalog no. 1610747; Bio-Rad) were added, and samples were boiled at 70°C for 10 minutes. Then, 40 µg of whole-cell extracts were fractionated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (8%) for 15 minutes at 120V and 60 minutes at 60V, and transferred to a polyvinylidene difluoride (catalog no. 1620177; Bio-Rad) membrane overnight at 30V. After incubation with 5% nonfat milk and 3% bovine serum albumin in Tris-buffered saline with Tween 20 (TBST) (10 mM Tris, pH 8.0, 150 mM NaCl, 1% Tween 20 (catalog no. P1379; MilliporeSigma) for 60 minutes, the membrane was washed once with TBST and incubated with antibodies against integrin β1 (D6S1W, catalog no. 34971T, Cell Signaling Technology), α-tubulin (11H10, catalog no. 21255; Cell Signaling Technology) at 4°C for overnight. Membranes were washed 3 times for 7 minutes and incubated with a 1:5000 dilution of horseradish peroxidase–conjugated anti-rabbit (catalog no. 7074S; Cell Signaling) antibodies for 2 hours. Blots were washed with TBST three times for 7 minutes and developed with the ECL Prime system (RPN2106, Amersham Biosciences) according to the manufacturer’s protocols.
Identification of master regulators in human leukemia
Gene regulatory networks were constructed by using ARACNe (Algorithm for the Reconstruction of Accurate Cellular Networks).38 For each human gene expression data set, the 5% of genes with the smallest variance were removed. Genes with regulatory functions were defined in Bioconductor data package “aracne.networks” (https://bioconductor.org). One hundred networks were constructed with random seeds and then consolidated as one consensus network. VIPER (Virtual Inference of Protein Activity by Enriched Regulon Analysis)39 was then used to identify potential master regulators (MRs) that drive activation of an LSC signature.40 A z score was used to quantify the enrichment level of this signature in every human sample within multiple AML data sets (GSE1159, GSE10358, GSE12417, GSE14468, GSE17855, and TCGA-LAML), as well as a recent pediatric study.41 The top 50 samples with the highest z scores were compared with the 50 samples with the lowest z scores. MRs were identified by using VIPER with an FDR of 5%.
Survival analysis
Survival analysis was performed with the Cox proportional hazards model using the “survival” package in R (R Foundation for Statistical Computing), comparing the survival of patients with the highest quartile of gene expression vs those with the lowest. Kaplan-Meier plots were used to visualize the association of FERMT3 with adverse prognosis, and the resulting P values were obtained with the log-rank test.
Results
In vivo CRISPR screening defines microenvironment-specific genetic dependencies of AML
To delineate the landscape of genes controlling AML pathobiology in vivo, we used 2 murine models that recapitulate the main clinical features observed in patients with AML. Model “MA” involves retroviral overexpression of the human fusion protein KMT2A/MLLT3 (MLL/AF9) in sorted murine multipotent progenitors, an event reported in ∼5% of adult and up to 20% of pediatric AML cases.30,42 Model “HM” involves retroviral overexpression of the transcription factors Hoxa9 and Meis1, which collaborate to promote leukemogenesis and are upregulated in AML downstream of specific genetic alterations, including KMT2A translocations, NPM1 mutations, and other poor-prognosis cytogenetic anomalies.31,43,44 Both HM and MA models were generated in hematopoietic progenitors isolated from Rosa26CAG-Cas9-GFP knock-in mice29 to ensure stable and robust expression of Cas9 (supplemental Figure 1A). When examining genome editing efficiency by disrupting the GFP cassette present in the knock-in cells, we observed a reduction of ∼90% of GFP-positive cells (supplemental Figure 1B), confirming high genome editing capacity in both models. As previously described,45 serial transplantation of MA and HM cells in vivo led to uniform and fast-growing murine AML models (supplemental Figure 1C).
We transduced both MA and HM cells with the lentiviral genome-wide mouse GeCKO v2 sgRNA library34 ex vivo before transplantation into sublethally irradiated mice. Bone marrow–derived cells were collected 14 days’ postinjection for next-generation sequencing analysis to monitor sgRNA representation (Figure 1A). Harvesting of AML cells 14 days’ posttransplantation ensured adequate recovery (50%-80%) of AML but occurred before development of clinically overt disease. Pooling of sequencing data from mice of the same experimental group (supplemental Figure 1D) led to the recovery of ∼80% of targeting sgRNAs and ∼90% of control sgRNAs (supplemental Figure 1E). Using the Wilcoxon rank sum test, we integrated the change in abundance of all sgRNAs targeting a given gene as a z score, which was compared with the z score of 6 control sgRNAs (supplemental Figure 1F). Using this approach, we evaluated the essentiality of 21 361 genes in the HM and MA models in both experimental contexts: in vitro only, in vivo only, or shared (Figure 1B-D; supplemental Table 1). As expected, genes known to be essential for bone marrow homing (Cxcr4) or integrin signaling (Fermt3 and Tln1) were specifically depleted in vivo (Figure 1C; supplemental Figure 1G). More generally, we observed that the majority of in vitro–only (77% [1157 of 1498]) and shared (87% [435 of 500]) gene sets we identified had previously been reported to be essential in other AML in vitro screens (Figure 1E).20,36,46 In contrast, only a small fraction of in vivo–only genes (37% [155 of 421]) had previously been identified. Because the MA model is based on a dysregulation of the H3Kme79-transferase DOT1L47 and Menin,48 a greater sensitivity of MA cells to the loss of Dot1l (MA z score, −3.74; HM z score, −1.46) and Men1 (MA z score, −3.83; HM z score, −1.66) was observed.
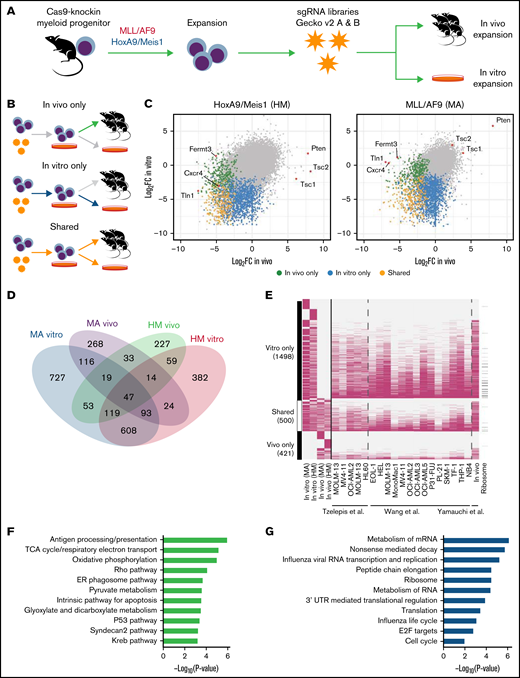
Genome-wide in vivo CRIPSR screens in 2 murine models of AML. (A) Schematic of the screening strategy. (B) Definition of 3 classes of statistically significant CRISPR hits according to time points compared. (C) For every gene screened, median log2FC of sgRNA representation, in vitro or in vivo as defined in panel B, for each cell line. (D) Numbers of genes that are essential (permutation P < .01) according to screening condition. (E) Overlap (pink cells) between genes identified as essential in our screen and other public screens. (F) Gene set enrichment analysis of genes identified to be essential only in vivo. (G) Gene set enrichment analysis of genes identified to be essential only in vitro or shared in vitro/in vivo. ER, endoplasmic reticulum; mRNA, messenger RNA; TCA, tricarboxylic acid; UTR, untranslated region.
Genome-wide in vivo CRIPSR screens in 2 murine models of AML. (A) Schematic of the screening strategy. (B) Definition of 3 classes of statistically significant CRISPR hits according to time points compared. (C) For every gene screened, median log2FC of sgRNA representation, in vitro or in vivo as defined in panel B, for each cell line. (D) Numbers of genes that are essential (permutation P < .01) according to screening condition. (E) Overlap (pink cells) between genes identified as essential in our screen and other public screens. (F) Gene set enrichment analysis of genes identified to be essential only in vivo. (G) Gene set enrichment analysis of genes identified to be essential only in vitro or shared in vitro/in vivo. ER, endoplasmic reticulum; mRNA, messenger RNA; TCA, tricarboxylic acid; UTR, untranslated region.
Gene set enrichment analysis confirmed that many of the in vivo–only genes are associated with antigen processing and presentation, metabolic pathways (tricarboxylic acid cycle, oxidative phosphorylation, and pyruvate metabolism), and Rho signaling (Figure 1F; supplemental Table 2), suggesting a unique dependency on these pathways for AML initiation and growth in vivo. As expected, in vitro–only and shared genes were enriched in essential biological processes, such as messenger RNA metabolism and translation (Figure 1G); examination of the genes depleted in both cell lines after 7 days of in vitro expansion also showed significant enrichment of key housekeeping cellular processes (supplemental Table 2). In summary, our genome-wide CRISPR screening approach identified key biological pathways that are essential to AML pathogenesis both in vitro and in vivo.
Validation of the genome-wide CRISPR screen using a custom targeted library
To validate genetic hits from the genome-wide screens, we selected a total of 1034 candidates based on their z score ranking, their lack of annotation as pan-essential, and their therapeutic potential (Figure 2A; supplemental Figure 2A). Briefly, we took the top-ranking genes depleted in vitro or in vivo, and removed known essential genes (https://github.com/macarthur-lab/gene_lists). In parallel, genes that were classified as drug targets, such as kinases and G protein–coupled receptors, were incorporated given their therapeutic potential (supplemental Table 3). We designed a custom lentiviral sgRNA library in which we incorporated 10 sgRNAs per gene as well as 1000 nontargeting sgRNA controls (total, 11 340 sgRNAs) and performed validation screening under the same experimental conditions as previously described (Figure 2B); the exception was that we used the algorithm MAGeCK37 to score each gene tested in our focused library (expressed as median log2-transformed change in sgRNA representation between time points; log2FC). As expected, we achieved greater coverage and recovery of the library in each biological replicate compared with the genome-wide approach (supplemental Figure 2B). For in vivo–depleted genes in MA or HM cell lines, >80% of the designed sgRNAs were depleted for 90% of the genes. Importantly, of 468 genes statistically depleted in vivo in the genome-wide screen, 365 were depleted in vivo in the validation library (FDR-adjusted P < .01), and 72 hits were specifically depleted in vivo (Figure 2C; supplemental Table 3). As expected, among top in vivo–only candidates (Figure 2D-E), we found genes that function as regulators of cellular interactions such as bone marrow homing (eg, the G protein–coupled receptor gene Cxcr4), immune response (B2m, Cr1l, Tap1, and Tap2), adhesion (Itgb1, Fermt3, and Tln1), cytoskeletal remodeling (Rock1), or suppression of phagocytosis (CD47).13 The in vivo context reproduced the effect of some known metabolic dependencies previously identified in vitro such as Pdxk25 (MA log2FC, −6.80; HM z score, −6.37).
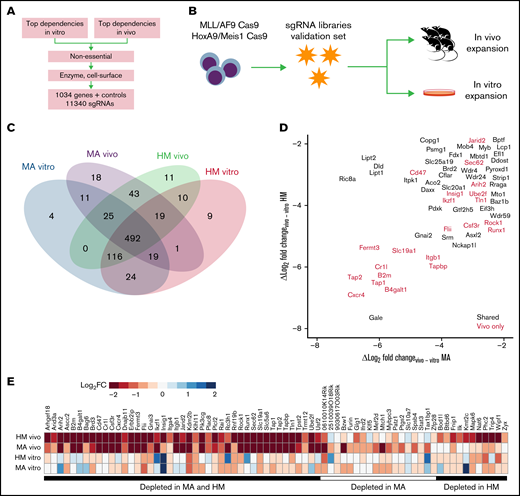
Validation of essential genetic hits using a focused CRISPR screen in vivo. (A) Schematic representation of the gene candidate selection workflow. (B) Schematic of the screening strategy. (C) Numbers of genes that are essential (P < .01) according to screening condition. (D) Specificity of the in vivo effect for every gene as represented by the log2FC in vivo depletion minus the in vitro depletion. Colors indicate genes that are essential (P < .01) according to screening condition. (E) Depletion values for the 72 genes that are essential in vivo (P < .01) for at least one cell line but not essential in vitro.
Validation of essential genetic hits using a focused CRISPR screen in vivo. (A) Schematic representation of the gene candidate selection workflow. (B) Schematic of the screening strategy. (C) Numbers of genes that are essential (P < .01) according to screening condition. (D) Specificity of the in vivo effect for every gene as represented by the log2FC in vivo depletion minus the in vitro depletion. Colors indicate genes that are essential (P < .01) according to screening condition. (E) Depletion values for the 72 genes that are essential in vivo (P < .01) for at least one cell line but not essential in vitro.
Immune suppressors and β-galactosylation are top in vivo–specific hits
To further understand the in vivo requirement for AML fitness, we examined in vivo–only hits that play a regulatory role or interact among themselves, as evidenced by STRING network (38 of 72 genes) (Figure 3A). Interestingly, among highly connected in vivo genes, we found the phosphoinositide 3-kinase catalytic subunit Pik3cg, an important mediator of extracellular signals and stromal interactions,49 the integrin intracellular partners Tln1 and Fermt3, and the G protein–coupled receptor inhibitory subunit Gnai3. Several in vivo–only hits participate in the immune response (GO_0002376) (Figure 3B; supplemental Table 4). Among these candidates, complement component receptor 1-like (Cr1l), which is a negative regulator of the complement system,50 scored highly in our in vivo screens, suggesting a requirement of this factor for suppression of complement attacks on AML cells in vivo.
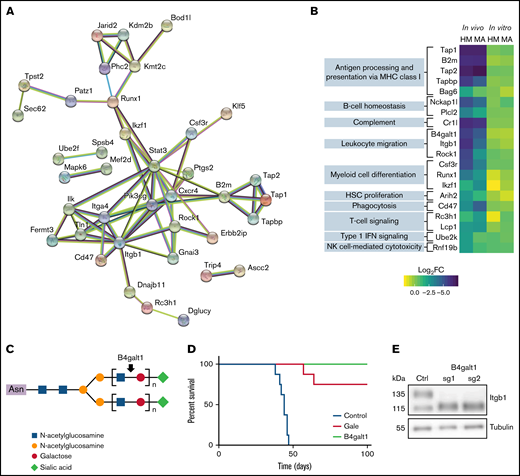
Essential role for β-galactosylation and immune regulators in vivo. (A) STRING functional protein association network of in vivo–specific hits with at least one connection. (B) For in vivo–only hits with gene ontology annotation “immune system process” (GO:0002376), log2FC depletion values according to condition and cell line. (C) Schematic representation of the galactosyltransferase role of B4galt1. (D) Overall survival of mice transplanted with MA cells after transduction with control sgRNA (sg Ctrl) or sgRNA targeting B4galt1 (2 distinct sgRNAs per group) (E) Immunoblot analysis of total lysates from control or B4galt1CRISPR-KO MA cells with an antibody against β1 integrin. HSC, hematopoietic stem cell; IFN, interferon; MHC, major histocompatibility complex; NK, natural killer.
Essential role for β-galactosylation and immune regulators in vivo. (A) STRING functional protein association network of in vivo–specific hits with at least one connection. (B) For in vivo–only hits with gene ontology annotation “immune system process” (GO:0002376), log2FC depletion values according to condition and cell line. (C) Schematic representation of the galactosyltransferase role of B4galt1. (D) Overall survival of mice transplanted with MA cells after transduction with control sgRNA (sg Ctrl) or sgRNA targeting B4galt1 (2 distinct sgRNAs per group) (E) Immunoblot analysis of total lysates from control or B4galt1CRISPR-KO MA cells with an antibody against β1 integrin. HSC, hematopoietic stem cell; IFN, interferon; MHC, major histocompatibility complex; NK, natural killer.
The β-galactosylation pathway members galactose epimerase (Gale) and β-1,4-galactosyltransferase-1 (B4galt1) emerged as key regulators of AML fitness in vivo in both our models (Figure 3C; supplemental Figure 3A). To confirm these genetic dependencies, CRISPR-mediated knockout MA cells were generated for both candidates and the loss of galactosylation validated at the cell surface by using RCA-I lectin staining (supplemental Figure 3C-D). Interestingly, loss of B4galt1 or Gale did not affect cell proliferation in vitro but prevented cell engraftment in vivo (Figure 3D). A previous report has shown that B4GALT1 glycosylates β1-integrin, which is critical for its role in thrombopoiesis and hematopoietic stem cell homeostasis.51 As expected, we noted a change in the electrophoretic mobility of β1-integrin in B4galt1 CRISPR-knockout MA cells (Figure 3E). Of note, Itgb1 itself is an essential gene for AML in vivo (HM log2FC, −5.93; MA log2FC, −4.09), suggesting that both β1-integrin and its posttranslational galactosylation are required for the engraftment of transplanted AML cells.
Identification of MRs of LSC fitness in human AML
To define the relevance of our candidates in human AML, we interrogated 7 distinct public AML data sets (GSE1159, GSE10358, GSE12417, GSE14468, GSE17855, TCGA, and a recent pediatric study41 ) for CRISPR hits that are important from a functional and prognostic standpoint. For each human AML gene expression data set, we constructed a global gene regulatory network using ARACNe38 and then inferred protein activity using VIPER39 of each MR in individual samples (Figure 4A). Specifically, because LSCs are an important therapeutic target in AML, we thus defined, in each data set, the top 50 samples with the highest enrichment of the LSC signature defined by Ng et al40 as one group and the lowest 50 samples as another group. We then identified MRs whose activity significantly differed between groups. Using this approach, a total of 1421 human AML-related MRs were identified with an FDR cutoff of 5%, and 820 of them were activated in at least 1 data set in samples with higher LSC signatures (supplemental Table 5).
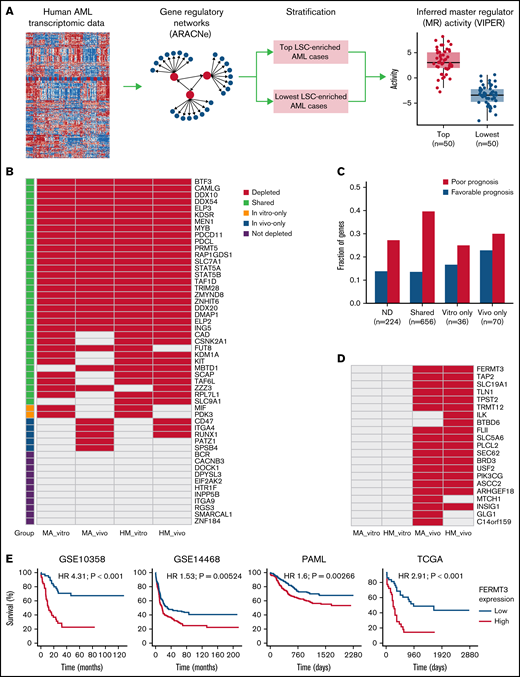
Integration of identified CRISPR hits and fitness signature to human AML. (A) Overview of computational approach. For the human AML gene expression data sets, a transcriptional regulatory network was constructed with ARACNe, and the protein activity of each MR in each sample was assessed by VIPER. Patient samples were stratified by enrichment of the LSC signature. The top 50 patients with highest enrichment were compared with those with lowest enrichment using VIPER to identify potential upstream MRs with an FDR cutoff of 5%. (B) MRs predicted to be activated in human AML samples with higher LSC, grouped according to whether they were homologs of CRISPR hits in vitro, in vivo, or in both conditions. Only homologs of genes that are covered in the secondary CRISPR screen are shown. (C) Prognostic analysis of CRISPR hits. The y-axis in the bar plot shows the fraction of human homologs of CRISPR hits that are significantly associated with survival in at least 1 of the 4 AML data sets (P values <.01), according to classification of CRISPR hits. (D) Homologs of in vivo–only CRISPR hits that are also associated with AML survival. (E) In each of the 4 AML data sets, patients were stratified by expression of FERMT3, and the top 25% samples with highest expression of FERMT3 were compared with the bottom 25%. Hazard ratios (HRs) and log-rank test P values are also shown.
Integration of identified CRISPR hits and fitness signature to human AML. (A) Overview of computational approach. For the human AML gene expression data sets, a transcriptional regulatory network was constructed with ARACNe, and the protein activity of each MR in each sample was assessed by VIPER. Patient samples were stratified by enrichment of the LSC signature. The top 50 patients with highest enrichment were compared with those with lowest enrichment using VIPER to identify potential upstream MRs with an FDR cutoff of 5%. (B) MRs predicted to be activated in human AML samples with higher LSC, grouped according to whether they were homologs of CRISPR hits in vitro, in vivo, or in both conditions. Only homologs of genes that are covered in the secondary CRISPR screen are shown. (C) Prognostic analysis of CRISPR hits. The y-axis in the bar plot shows the fraction of human homologs of CRISPR hits that are significantly associated with survival in at least 1 of the 4 AML data sets (P values <.01), according to classification of CRISPR hits. (D) Homologs of in vivo–only CRISPR hits that are also associated with AML survival. (E) In each of the 4 AML data sets, patients were stratified by expression of FERMT3, and the top 25% samples with highest expression of FERMT3 were compared with the bottom 25%. Hazard ratios (HRs) and log-rank test P values are also shown.
Eighty-one of these MRs were tested in the secondary CRISPR screens, and 52 of them were activated in at least 1 of the data sets tested (Figure 4B). We found that 79% (41) of activated MRs were depleted either in vitro or in vivo, with only 3 in vivo–specific hits shared between both models (CD47, ITGA4, and RUNX1). As previously discussed, CD47 is a known suppressor of innate immune response and potential therapeutic target, and ITGA4 mediates cell adhesion. We found, in both MA and HM models, that Runx1 was only essential in vivo and not in vitro. Importantly, our in vivo screen could reproduce the in vitro essentiality of most LSC-associated MRs, and our approach highlighted several hits currently investigated in clinical trials (eg, MEN1 and PRMT5). These data suggest that our CRISPR-based computational predictions successfully identified functionally relevant MRs of AML.
In parallel, we interrogated the clinical relevance of our hits by correlating them to patient outcome in 4 distinct AML cohorts (GSE10358, GSE14468, PAML, and TCGA) (Figure 4C; supplemental Table 6). In total, 8 in vivo–specific hits were identified whose expression was associated with adverse prognosis in at least 2 of the 4 cohorts tested (BTBD6, FERMT3, ILK, SLC19A1, TAP2, TLN1, TPST2, and TRMT12) (Figure 4D). FERMT3 was a CRISPR hit depleted only in vivo and strongly associated with shorter survival in the 4 AML data sets (Figure 4E). Together, these data highlight the power of using parallel CRISPR screens to identify essential MRs of AML growth as well as prognostic factors for AML.
Discussion
AML arises from cooperating genetic changes leading to aberrant self-renewal and increased proliferation of myeloid progenitors.52-56 Importantly, leukemic cells benefit from the supportive bone marrow microenvironment to proliferate and be resistant to therapy.9 To detect genes that are essential to the development of AML in vivo, we developed a genome-wide CRISPR screening pipeline in 2 murine models with highly penetrant genetic drivers: KMT2A/MLLT3 and Hoxa9/Meis1.30,31 It is important to note that CRISPR targeting was done in AML cells before injection, and thus our screening results incorporate genes that are relevant to leukemic homing to the bone marrow.
Our in vivo screens revealed a distinct set of metabolic, migratory, and immune factors preferentially and specifically depleted in vivo. For example, Cd47 is a known suppressor of phagocytosis and therapeutic target in hematologic malignancies.13 Loss of several components of the major histocompatibility complex I (Tap1, Tap2, Tapbp, and B2m) impaired AML engraftment and/or growth. We interpreted this result as loss of a major inhibitory signal directed toward host-derived natural killer cells, leading to immune clearance. Likewise, loss of a negative regulator of complement activation (Cr1l, also known as Crry) led to reduction of AML proliferation. Cr1l is highly expressed in the hematopoietic system and prevents complement activation on hematopoietic cells.50,57 Expression of several members of the complement pathway, including CFD, CFH, and SERPING1, have been previously linked to AML patient outcome,58 highlighting the potential contribution of this pathway in the pathobiology of AML.
β-1,4-galactosylation links uridine diphosphate galactose (UDP-galactose) metabolism and the posttranslational modifications of cell surface proteins relevant for AML migration and immune regulation.59,60 Gale is an essential enzyme for the conversion of UDP-glucose to UDP-galactose, thereby providing an essential metabolite for all galactosylation reactions within the organism, whereas B4galt1 mediates a more terminal modification of carbohydrate chains. B4galt1 and Gale were preferentially depleted in our in vivo screens, and we show that B4galt1 knockout AML cells cannot engraft, likely due to the altered posttranslational status of integrin β1, which has recently been linked to hematopoietic stem cell engraftment and homeostasis.51 More generally, variations in N-glycosylation of proteins have been linked to several immunologic and hematologic disorders,61 and their precise contribution to AML initiation and progression remains an active area of research. Of note, although this analysis was focused on identifying essential genes for AML growth in the bone marrow (the primary site of disease in humans), the experimental approach outlined herein could be applied to different tissues involved by AML (eg, spleen and liver) in future studies.
To correlate our screening results to human AML, we performed a network-based analysis of human genes inferred to be MRs of AML LSCs and prioritized essential CRISPR hits according to their regulatory function. This list of 41 MRs includes targets currently investigated clinically such as PRMT5 and MEN1, suggesting that both approaches are efficient to define the essential genetic regulators of AML pathogenesis. We observed that the transcription factor RUNX1 was not essential in vitro in our models, in agreement with a prior report62 and public screening data (Broad Institute Cancer Dependency Map) for MLL-AF9–driven AML. However, we observed a strong in vivo dependency, suggesting that RUNX1 may regulate genes that are essential for in vivo interactions. We observed the critical role of integrin signaling in AML through essentiality of both the signaling adapters FERMT3 and TLN1 and the direct implication of B4GALT1 that regulates β1 integrin.51 FERMT3 and TLN1 expression correlate with a poorer prognosis in human AML patients, along with other recent reports of dysregulated integrin signaling in high-risk AML.63 Although direct therapeutic inhibition of FERMT3 and TLN1 in patients with AML may be complicated due to their essential role in leukocyte migration and platelet function,64,65 further investigation of the intracellular signaling events mediated by FERMT3 and TLN1 in AML cells in vivo might identify therapeutic vulnerabilities that are specific to AML. It is possible that these interactions facilitate the resistance to treatment, something that could be investigated using similar in vivo screening approaches.
Overall, our CRISPR screens provide a computational pipeline and genome-wide map of differential genetic dependencies in vivo and in vitro, a precious resource for any investigator hoping to better understand the microenvironmental interactions of AML and their impact on patient outcomes. Because resistance of AML to chemotherapy is thought to be mediated in part by microenvironmental interactions that promote metabolic rewiring, cell quiescence, and pro-survival signaling,9 the in vivo genetic dependencies defined in this screen whose overexpression adversely affect patient survival may represent drivers of such interactions that could be investigated in future mechanistic studies.
Acknowledgments
F.E.M. is a recipient of a Clinician-Scientist Award from the Fonds de Recherche du Québec-Santé. J.S. is a recipient of the Hundred Talents Program Award of the Chinese Academy of Sciences. M.J. is a recipient of a Cole Foundation doctoral scholarship and Lady Davis Institute/TD Bank Studentship Award. Work in the F.E.M. laboratory was supported by an operating grant from the Cancer Research Society (#23604) and grants from the Cole Foundation, Richard and Edith Strauss Foundation, and Jewish General Hospital Foundation. This work was supported by the Dana-Farber Cancer Institute Physical Science Oncology Center (National Institutes of Health, National Institute of Cancer, U54CA193461).
Authorship
Contribution: F.E.M. and J.S. designed, performed most of the functional and computational validation experiments, analyzed the data, and wrote the manuscript; D.B.S., T.O., M.J., C.H.M., Y.S.K., E.M., A.Z., and R.V. performed functional validation experiments presented in the manuscript and analyzed the data; S.H. performed additional computational analysis presented in the manuscript; J.D. assisted with experimental design and analysis of the CRISPR screens; A.O. contributed to the analysis and interpretation of the data; and F.M. and D.T.S. conceived the study, designed the research, provided supervision, and wrote the manuscript with input from all the other authors.
Conflict-of-interest disclosure: D.T.S. is a director and stockholder of Magenta Therapeutics, Agios Pharmaceuticals, Editas Medicine, Clear Creek Bio, and LifeVaultBio; a founder and stockholder of Fate Therapeutics; a consultant for FOG Pharma, Inzen, and VCanBio; and receives research support from Dainippon Sumitomo Pharma. D.B.S. is a cofounder and holds equity in Clear Creek Bio; is a consultant and holds equity in SAFI Biosolutions; and is a consultant for Keros Therapeutics. F.M. is a cofounder of and has equity in Harbinger Health; has equity in Zephyr AI; and serves as a consultant for Harbinger Health, Zephyr AI, and Red Cell Partners. The remaining authors declare no competing financial interests.
The current affiliation for F.E.M. is Division of Experimental Medicine and Division of Hematology, Department of Medicine, McGill University, Montreal, QC, Canada; and Lady Davis Institute for Medical Research, Jewish General Hospital, Montreal, QC, Canada.
The current affiliation for J.S. is State Key Laboratory of Molecular Biology, Shanghai Institute of Biochemistry and Cell Biology, Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai, China.
The current affiliation for T.O. is Takeda Pharmaceuticals.
The current affiliation for Y.S.K. is Moderna Therapeutics.
Correspondence: Francois E. Mercier, Lady Davis Institute for Medical Research, 3755 Côte-Sainte-Catherine Rd, Montréal, QC H3T 1E2, Canada; e-mail: francois.mercier@mcgill.ca; or Franziska Michor, Dana-Farber Cancer Institute, CLS Building, 3 Blackfan Circle, Boston, MA 02115; e-mail: michor@jimmy.harvard.edu; or David T. Scadden, Massachusetts General Hospital, Simches Research Building, 185 Cambridge St, Boston, MA 02114; e-mail: david_scadden@harvard.edu.

