Abstract
Myelodysplastic syndrome (MDS) typically presents in older adults with the acquisition of age-related somatic mutations, whereas MDS presenting in children and younger adults is more frequently associated with germline genetic predisposition. Germline predisposition is increasingly recognized in MDS presenting at older ages as well. Although each individual genetic disorder is rare, as a group, the genetic MDS disorders account for a significant subset of MDS in children and young adults. Because many patients lack overt syndromic features, genetic testing plays an important role in the diagnostic evaluation. This review provides an overview of syndromes associated with genetic predisposition to MDS, discusses implications for clinical evaluation and management, and explores scientific insights gleaned from the study of MDS predisposition syndromes. The effects of germline genetic context on the selective pressures driving somatic clonal evolution are explored. Elucidation of the molecular and genetic pathways driving clonal evolution may inform surveillance and risk stratification, and may lead to the development of novel therapeutic strategies.
Introduction
Myelodysplastic syndrome (MDS) is defined by the World Health Organization as a clonal hematopoietic disorder characterized by ineffective hematopoiesis, cytopenias, single- or multilineage dysplasia, and an increased propensity to evolve to acute myeloid leukemia (AML).1 Primary MDS typically presents in older adults, with median age of disease onset of 72 to 75 years in the context of somatic mutations acquired with age.2,3 MDS presenting in children and younger adults is more frequently associated with germline genetic predisposition.4 In recognition of the growing subgroup of MDS or acute leukemia associated with germline mutations or familial cases, the revised World Health Organization classification of myeloid neoplasms now includes a new category: myeloid neoplasms with germline predisposition.1
A high risk for MDS in childhood and early adulthood is characteristic of certain genetic bone marrow failure syndromes.5 Although physical stigmata and family history provide important clues to an underlying genetic MDS predisposition syndrome, it is now clear that clinically silent phenotypes with cryptic presentations are common. Several genomic studies demonstrated that a subset of pediatric and young adult patients younger than 40 years presenting with MDS have unrecognized bone marrow failure and genetic MDS syndromes.4,6,7 Although many published studies on germline genetic predisposition thus far have typically used the adolescent and young adult upper age range of 40 years, it is critical to note that genetic MDS predisposition syndromes also present in older adulthood. In particular, germline mutations in DDX41 present with myeloid malignancies in older adults. Germline telomere biology disorders, GATA2, thrombocytopenia-associated disorders, and others may present across all ages. Older age at presentation is insufficient to rule out a germline predisposition disorder. Indeed, before the availability of genetic testing, diagnosis of MDS predisposition was skewed toward patients presenting with overt syndromic features in childhood, whereas nonsyndromic patients presenting with MDS in adulthood were previously missed. Additional data are needed to inform evidence-based determination of age of presentation for each MDS predisposition disorder in the era of genetic testing. Recent studies have shown that although each individual genetic disorder is rare, as a group they account for approximately 4% to 15% of patients with MDS.4,6,7 These findings elucidate the broad phenotypic spectrum of these genetic syndromes and highlight the importance of screening for these disorders.
The diagnosis of a germline MDS predisposition disorder carries profound implications for medical management and treatment. Baseline dysplasia associated with some MDS predisposition disorders must not be confused with frank MDS. Prompt diagnosis of these disorders allows surveillance to detect early signs of disease progression and timely hematopoietic stem cell transplant (HSCT) before progression to leukemia, which often carries a poor prognosis. Many of these genes are associated with additional medical complications or comorbidities that may require modified conditioning regimens for HSCT. Genetic testing of potential family member donors is essential before HSCT to avoid choosing a donor carrying the same germline mutations as the proband. Diagnosis also allows family counseling.
This review provides an overview of genetic predisposition to MDS, discusses clinical evaluation and management, and explores scientific insights garnered from the study of MDS germline genetics. The germline genetic MDS disorders advance our understanding of the genes that govern hematopoiesis, as well as the cellular and molecular perturbations that lead to malignant transformation. Last, we explore the emerging hypothesis that somatic mutations may result in hematopoietic stem cell adaptation or oncogenic transformation driven by specific selective pressures exerted by the germline genetic context of the hematopoietic stem cell. Because of space limitations, only a subset of disorders in which MDS predisposition is a predominant feature are included here. This focused review of MDS predisposition does not review the broader classification of leukemia predisposition disorders such as lymphoid malignancies, myeloproliferative syndromes, chronic myelomonocytic leukemia, or RASopathies, so the reader is referred to the other articles within this issue, as well as excellent recent reviews of these topics and the references therein.8-11
Germline MDS syndromes
GATA2
GATA2 encodes a zinc finger transcription factor critical for hematopoiesis, hematopoietic stem cell homeostasis, and lymphatic development (see Spinner et al12 and McReynolds et al13 and references therein). The phenotypic spectrum of germline GATA2 mutations is broad and may present with MonoMac syndrome14 or Emberger syndrome15 (Table 1). However, germline GATA2 mutations may also present with isolated neutropenia or bone marrow failure without syndromic features or family history.16-18 The bone marrow histology in GATA2 deficiency is typically hypocellular and may manifest characteristic megakaryocyte dysmorphologies with micronuclei or splayed nuclei19 (Figure 1). Additional findings include monocytopenia and immunologic abnormalities.
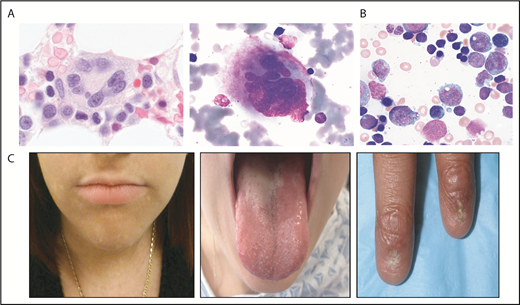
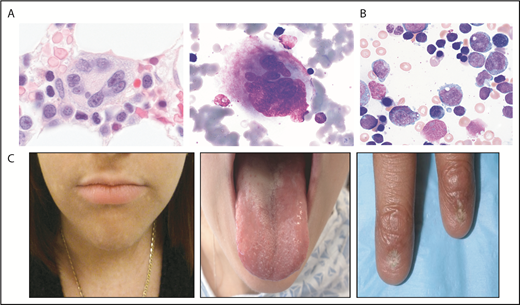
Selected bone marrow histology and physical findings associated with genetic MDS predisposition syndromes. (A) Hematoxylin and eosin (left panel) or May-Grunwald Giemsa staining of abnormal bone marrow megakaryocytes present in GATA2 predisposition disorders. (B) May-Grunwald Giemsa staining of bone marrow aspirate from a patient with SCN demonstrating myeloid maturation arrest (marrow images courtesy of M. Fleming). (C) Patient features characteristic of diagnostic triad of TBDs, including lacy/reticular pigmentation on neck and trunk, oral leukoplakia, and dystrophic fingernails (images courtesy of S. Agarwal).
Selected bone marrow histology and physical findings associated with genetic MDS predisposition syndromes. (A) Hematoxylin and eosin (left panel) or May-Grunwald Giemsa staining of abnormal bone marrow megakaryocytes present in GATA2 predisposition disorders. (B) May-Grunwald Giemsa staining of bone marrow aspirate from a patient with SCN demonstrating myeloid maturation arrest (marrow images courtesy of M. Fleming). (C) Patient features characteristic of diagnostic triad of TBDs, including lacy/reticular pigmentation on neck and trunk, oral leukoplakia, and dystrophic fingernails (images courtesy of S. Agarwal).
MDS with germline GATA2 mutations is frequently associated with monosomy 7/del7q (-7) or trisomy 8, particularly in children and younger adults.20,21 A study of 426 cases of pediatric MDS identified germline GATA2 mutations in 37% of patients with primary MDS with -7 and 16% of MDS cases with trisomy 8.20 Germline GATA2 mutations were identified in 72% of adolescents with -7 MDS. In contrast, germline GATA2 mutations were absent in therapy-related MDS.
SAMD9/SAMD9L
Initial reports associating refractory anemia, monosomy 7, and AML were described by E.J. Freireich in the 1960s.23 Subsequent studies of the recurrent common microdeletion region at 7q22 led to discovery of a region containing 2 genes harboring the sterile α motif (SAM) domain-9 (SAMD9) and SAMD9L (SAMD9-like; see Wlodarski et al21 and Davidsson et al24 and references therein).
Homozygous germline loss-of-function mutations in SAMD9L are associated with normophosphatemic familial tumoral calcinosis,25 whereas heterozygous gain-of-function missense mutations in SAMD9 or SAMD9L are associated with cytopenias and a high risk of developing MDS with monosomy 7/del7q, with variable additional clinical phenotypes (Table 1). Germline mutations in SAMD9 and SAMD9L were initially identified in MIRAGE syndrome26 and ataxia-pancytopenia syndrome,27,28 respectively (Table 1). Patients frequently present with bone marrow failure and a high risk for monosomy 7 MDS at a young age.29-31 Patients with germline SAMD9 and SAMD9L may present with MDS or AML without syndromic features.
The molecular functions of these genes remain poorly understood; both genes exert negative effects on cell proliferation consistent with tumor suppressors.28,31 SAMD9/SAMD9L share a common evolutionary history and encode proteins with 60% homology. Murine studies indicate that SAMD9L functions as an endosomal fusion facilitator and, in the haploinsufficient state, leads to MDS.32 Studies of clonal evolution in these disorders are discussed here.
Fanconi anemia
A century ago, Swiss pediatrician Guido Fanconi described a family with 3 sons with various physical abnormalities and pernicious anemia.33 Now known as Fanconi anemia (FA), this disorder is characterized by genomic instability, hypersensitivity to DNA cross-linking agents, and predisposition to marrow failure, as well as hematologic malignancies and solid tumors.34-36 The congenital anomalies variably associated with FA are numerous (Table 1). A significant subset (25%-30%) of patients lack these physical findings.37 Hematologic abnormalities are variable and include cytopenias, red cell macrocytosis, hypocellular marrow with mild dysplasia, and bone marrow failure with increased risk for MDS or AML. The cumulative incidence for MDS was 40% by age 50 years and 20% by age 40 years for AML.34 The incidence of leukemia is even higher in the FANCD1/BRCA2 subtype of FA, with many cases presenting at younger than 5 years.38
This clinically and genetically diverse syndrome is caused by germline mutations in any of at least 23 FA genes (FANCA-FANCW) that function coordinately in DNA repair.39 Cells from patients with FA manifest increased chromosomal breakage and radial formation after exposure to mitomycin C or diepoxybutane.40,41
Of particular note in FA is the importance of reduced-intensity conditioning for HSCT because of the heightened toxicity of radiation and alkylator-based conditioning regimens in these patients.42,43 Unlike other MDS syndromes that can be cured by HSCT, these patients have increased morbidity posttransplantation and have a higher risk for solid tumors compared with nontransplanted patients.44-46
Diamond-Blackfan anemia
Diamond-Blackfan anemia (DBA) was first described by Diamond and Blackfan in the 1930s as a congenital hypoplastic anemia.47 DBA registries across the world are elucidating the clinical and molecular features of DBA (see Da Costa et al,48 Vlachos et al,49 and Vlachos et al50 and references therein) Approximately half of patients with DBA exhibit physical abnormalities (Table 1). The majority of DBA cases are associated with increased levels of erythrocyte adenosine deaminase activity.51 Patients typically present in infancy with macrocytic anemia and reticulocytopenia. The bone marrow histopathology typically shows red cell aplasia in an otherwise normocellular marrow. Major causes of morbidity and mortality are related to treatment adverse effects and long-term risks for malignancy, including MDS and solid tumors.52,53 In a study from the DBA Registry of North America, 2% of patients with DBA had developed AML by age 45 years.52
DBA is most frequently caused by autosomal dominant mutations, resulting in haploinsufficiency of genes encoding ribosomal proteins, most commonly RPS19.54 Around 50% of ribosomal gene mutations arise de novo.48 X-linked mutations in GATA1, which encodes a transcription factor critical for erythropoiesis, also cause DBA.55 X-linked mutations in TSR2 that binds RPS26 have also been reported. How mutations in ubiquitously expressed genes encoding an essential organelle result in tissue-specific phenotypes has been a subject of intense interest. Disease mechanisms include p53-mediated apoptosis induced by ribosomal stress,56 increased cell death from free excess heme with delayed globin production,57 increased autophagy,58 and altered translation of select erythroid-specific transcripts such as GATA1.59,60 In adult 5q- syndrome MDS, an acquired deletion resulting in haploinsufficiency of the RPS14 gene is associated with MDS61 and sensitivity to lenalidomide.62
Dyskeratosis congenita/telomere biology disorders
Dyskeratosis congenita (DC) or telomere biology disorders (TBDs) encompass genetically heterogeneous disorders related to impaired telomere maintenance (see Townsley et al,63 Bertuch et al,64 and Agarwal et al65 and references therein). These deficiencies lead to a broad and variable clinical phenotype (Table 1; Figure 1), but patients often present without overt syndromic features.66 DC/TBDs are frequently associated with hematologic complications including bone marrow failure, MDS, and AML.65,67 The actuarial risk (absent competing risks) of MDS in the patients with DC reached a plateau of 3% at 29 years of age.68 The cumulative incidence of MDS in DC has been estimated to be 2% by age 50 years.69 MDS may be the sole presenting clinical feature. Screening for TBDs involves evaluation of lymphocyte telomere lengths, which are typically below the first percentile for age across multiple lymphocyte subsets.70,71 The sensitivity of this test may be lower in adults as a result of decreased telomere lengths with increasing age. DC/TBD is associated with many extrahematologic complications, particularly pulmonary fibrosis, hepatic abnormalities, and vascular anomalies.72,73 Patients are sensitive to toxicities from chemotherapy and radiation and warrant specially tailored transplant regimens.65,74,75
DC/TBDs are caused by mutations in at least 13 genes involved in telomere maintenance with variable inheritance patterns (Table1).64 Genetic testing is helpful diagnostically, as shortened telomeres are sometimes noted in other disorders.71 Telomeres are specialized protein:DNA complexes protecting chromosome ends that prevent genomic instability resulting from free DNA ends (see Townsley et al63 and references therein). Telomeres shorten over progressive cycles of DNA replication. Critically short telomere lengths trigger senescence and cell death.
Shwachman Diamond syndrome
In 1964, Shwachman and Diamond observed children in a cystic fibrosis clinic who had pancreatic insufficiency and neutropenia and normal sweat tests, without pulmonary manifestations.76 Separately, Bodian described cases of familial exocrine pancreatic insufficiency.77 Shwachman Diamond syndrome (SDS) is characterized by exocrine pancreatic dysfunction and bone marrow failure, as well as additional physical findings (Table 1). However, these features are not present in all patients,78 and patients may present with MDS as the first observed feature of disease.4 The most common hematologic abnormality is neutropenia, which may be mild or severe and may be transient or persistent. Additional hematologic complications include bone marrow failure, MDS, and AML. In a French cohort of 102 patients with SDS, the cumulative incidence of MDS/AML was 18.8% at 20 and 36.1% at 30 years of age.79 The bone marrow is typically hypocellular for age, with mild dysgranulopoietic features. The most sensitive test of exocrine pancreatic dysfunction in SDS is serum trypsinogen below age 3 years or pancreatic isoamylase at or above age 3 years.80
SDS is most commonly caused by autosomal recessive mutations in the eponymous SBDS gene, leading to low levels of SDS protein.81 SDS is involved in the joining of the large (60S) and small (40S) ribosomal subunits and functions as a cofactor for elongation factor-like (EFL1) to remove the antiassociation factor eIF6 from the large (60S) subunit.82-85 SDS also plays a role in mitotic spindle stabilization.86 Data from the North American SDS Registry reveal a spectrum of SBDS mutations including missense, splice site, nonsense, frameshift, and partial or whole gene deletions. Recently, autosomal recessive mutations in DNAJC2187,88 and EFL1,89 which also function in ribosome maturation, have been found in patients with variable clinical features of SDS. Autosomal dominant mutations in SRP54 have also been found in patients with SDS-like features.90 SRP54 protein is a component of the ribonucleoprotein complex that mediates cotranslational targeting of secretory and membrane proteins to the endoplasmic reticulum. Patients with SRP54 mutations may have exocrine pancreatic insufficiency or severe neutropenia as a presenting symptom.91 Thus far, AML has been described in a patient with DNAJC21 mutations,90 and no cases of MDS or AML have been described in patients with EFL1 or SRP54 mutations.
Familial MDS associated with thrombocytopenia
RUNX1
RUNX1 deficiency is the cause of an autosomal dominant familial platelet disorder with predisposition to myeloid leukemia (see Bellissimo et al92 and Schlegelberger et al93 and references therein). Familial platelet disorder with predisposition to myeloid leukemia typically presents with mild to moderate thrombocytopenia with normal-sized platelets, functional platelet defects leading to excessive bleeding, and an increased risk of developing MDS, AML, or lymphoid malignancies such as T-cell acute lymphoblastic leukemia (T-ALL) or B-cell malignancies. The thrombocytopenia may be mild and asymptomatic. Qualitative platelet abnormalities may cause bleeding in excess of that predicted by the platelet count. Although longitudinal data are limited, lifetime risk for myeloid malignancy in patients with germline RUNX1 mutations appears to be highly variable and has been estimated to be around 44%, but ranges from 11% to 100% (reviewed in Godley et al22 ). Disease anticipation whereby the clinical manifestations arise at younger ages in subsequent generations has been described in families with germline RUNX1 mutations.
RUNX1 encodes a heterodimeric transcription factor essential for hematopoiesis, megakaryopoiesis, and platelet function. RUNX1 functions as a transcriptional activator for some genes and a transcriptional repressor for others. Germline mutations in RUNX1 include missense, nonsense, frameshift, duplication, partial or full gene deletion mutations, or gene rearrangements.22 Missense point mutations often occur in the highly conserved RUNT homology domain at sites affecting DNA binding or heterodimerization. Truncations within the RUNT domain may result in hypomorphic mutations.
ETV6
Autosomal dominant germline mutations in ETV6 (also known as TEL) are associated with inherited thrombocytopenia and increase risk for both myeloid and lymphoid hematologic malignancies (see Hock et al94 and references therein). Clinical features of germline ETV6 mutations include thrombocytopenia, platelet function abnormalities, macrocytosis, and increased bleeding tendency. The marrow may be hypocellular with dysmegakaryopoiesis, variable megakaryocyte numbers, and eosinophilia.95 Germline ETV6 mutations are predominantly found in ALL but are also reported in myeloid malignancies including MDS and AML.96-101 Data are scant to inform disease penetrance. Thus far, the numbers of total patients reported is too few to ascertain associated syndromic features.
ETV6 encodes a transcriptional repressor critical for hematopoiesis, megakaryopoiesis, and embryonic development.94 Germline ETV6 mutations are typically located in the DNA binding ETS domain and result in autosomal dominant inhibition of ETV6 function through dimerization. A P214L mutation located in the linker region implicated in release of an autoinhibitory domain has also been found in multiple unrelated kindreds.
ANKRD26
An autosomal dominant disorder characterized by quantitative and qualitative platelet disorders and increased risk for MDS and AML is also caused by mutations in ANKRD26 (see Noris et al102 and references therein). This disorder is characterized by moderate thrombocytopenia with normal platelet size, increased bleeding, and abnormal platelet function and aggregation.103,104 Bone marrow exam is notable for megakaryocyte dysplasia with small megakaryocytes containing hypolobated nuclei. Thus far, of the reported cases, about 5% developed acute leukemia (2.2% had MDS and 1.3% had CML), which is notably higher than expected in the general population.102
ANKRD26 encodes a protein with N-terminal ankyrin repeat domains believed to function in protein-protein interactions. Although the function of ANKRD26 is unknown, expression profiling demonstrates a presence in megakaryocytes, and less so in erythroid cells.105 Mutations in ANRK26 are typically point mutations located in the 5′ untranslated region of the gene, although deletions and point mutations within the coding region have been reported.22 The 5′UTR mutations impair the binding of repressive transcription factors such as RUNX1 and FLI1 to this regulatory region, resulting in aberrantly increased ANKRD26 expression, which in turn disrupts platelet production.106
DDX41
Germline mutations in DDX41 have been associated with autosomal dominant familial MDS/AML presenting in older age.107-110 Patients typically present with an increased risk for normal karyotype MDS or AML. Additional hematologic malignancies have also been described (Table 1). In comparison with other germline MDS syndromes, those associated with DDX41 mutation present at older age. The bone marrow is typically hypocellular. Some cases with antecedent cytopenias and macrocytosis preceding presentation with myeloid malignancy have been reported. No common nonhematologic phenotype has been associated with DDX41 germline mutations, and family history is often the sole clinical clue to DDX41 germline predisposition. DDX41 encodes a helicase possibly functioning in RNA splicing. Germline mutations in DDX41 include frameshift, missense, nonsense, and whole-gene deletion/contiguous gene mutations.111 In contrast, somatic mutations associated with malignancy are typically missense mutations.
SRP72
Germline mutations in the ribonuclear protein complex gene SRP72 have been described with autosomal-dominant MDS and bone marrow failure. Two families were found to have heterozygous mutations in SRP72 associated with autosomal dominant aplastic anemia, and the patients with MDS presented in their third and fifth decade of life.112 The SRP72 gene encodes a subunit of the signal recognition particle complex that is responsible for the trafficking to the endoplasmic reticulum. Mutations thus far described are frameshift and missense mutations.112
ELANE
Severe congenital neutropenia (SCN) is a genetically heterogeneous disorder characterized by neutropenia typically less the 500/μL (see Skokowa et al113 and references therein). SCN is caused by mutations in several different genes, most commonly ELANE, which encodes neutrophil elastase114 (NE; Table 1). These patients usually present in early infancy with severe neutropenia and infectious complications, although an antecedent normal neutrophil count does not rule out a genetic neutropenia disorder. Bone marrow findings with ELANE mutations include maturation arrest of neutrophil precursors at the promyelocyte stage in the bone marrow (Figure 1).115 Overall cumulative incidence of sepsis death in SCN is 10%, and 22% for development of MDS/AML after 15 years of treatment with granulocyte colony-stimulating factor (G-CSF).116 The overall hazard of MDS/AML is 2.3%/year after 10 years receiving G-CSF.116 A poor absolute neutrophil count response despite high doses of G-CSF is associated with an increased risk for development of MDS/AML likely related to disease severity.116 Mutations are scattered throughout the ELANE gene and include missense, nonsense, frameshift, splice, insertion, and deletion mutations.117
The mechanism by which ELANE mutations lead to the phenotype of SCN is not well understood. Data support multiple etiologies for hematopoietic cellular stressors such as misfolding of NE protein leading to activation of the unfolded protein response,118,119 mislocalized NE,120 or altered substrates of mutant NE protein121 with increased apoptosis.
Li Fraumeni Syndrome/TP53
First described by Frederick Li and Joseph Fraumeni, this autosomal-dominant disorder is characterized by a high risk for a spectrum of solid tumors and hematologic malignancies at a young age. Germline mutations in TP53 are associated with Li Fraumeni syndrome (LFS). The tumor spectrum includes soft tissue sarcomas, premenopausal breast cancer, central nervous system (CNS) tumors, adrenocortical carcinomas, pancreatic tumors, and others, as well as MDS and lymphoid or myeloid malignancies (see Quinn et al,9 Guha et al,122 and Kratz et al123 and references therein). LFS is highly penetrant, with a cumulative cancer incidence, mostly of solid tumors, of 50% by age 31 years for females and 50% by age 46 years for males, and nearly 100% by age 70 years for either sex.124 Leukemia, typically hypodiploid ALL but also AML and CML, occur in around 2% to 4% of patients with LFS.122 MDS in patients with LFS tends to arise secondary to treatment of another primary neoplasm, although primary MDS is also seen. The p53 protein plays a central role in checkpoint controls of cell growth, survival, and stress responses involving cell cycle arrest, apoptosis, senescence, DNA repair, and metabolic stressors. Missense mutations are the most common type of germline TP53 mutation (74%), often located within exons 5 to 8 in the cord DNA-binding domain, and may result in either dominant-negative or loss-of-function mutations. Deletions involving all or part of TP53, nonsense mutations, and splice site mutations are also found.
Diagnosis of genetic predisposition to MDS
Genetic predisposition to MDS should be considered in a patient presenting at a young age with bone marrow failure, MDS, or AML, or with unexpected hematologic toxicity during treatment of a malignancy at a young age. Clinical clues include physical anomalies, endocrinopathies, short stature or failure to thrive, or immune deficiencies in a patient with hematologic abnormalities such as cytopenias, unexplained red cell macrocytosis, or frank malignancy (Table 1). Family history of first- or second-degree relatives with malignancy, cytopenias, congenital anomalies, or excessive toxicity with chemotherapy or radiation also raise concern for a genetic MDS predisposition disorder. The lack of characteristic clinical features or negative family history does not exclude the presence of a germline MDS syndrome. Germline mutations can arise de novo or may result from parental gonadal mosaicism. In addition, several of these germline MDS syndromes have no overt syndromic features or have variable penetrance or delayed expressivity. Cytogenetic clonal abnormalities common to certain genetic MDS disorders may prompt further investigation. For example, MDS with monosomy 7 frequently arises in patients with germline mutations in GATA2, SAMD9, SAMD9L, or genetic bone marrow failure syndromes.125 Del20q or isochromosome 7 are common in SDS.126 1q+ and 3q26q29 amplifications are common in FA.127-129
Patients and families with suspected germline genetic MDS syndromes should receive genetic counseling regarding the indication for sending genetic testing, limitations of such testing, and family counseling. The strengths and weaknesses of different sequencing options were recently discussed.130 The clinician should be aware of which genes are included and whether all regions of interest within those genes are covered by the genetic tests ordered. With features noted previously or a strong clinical suspicion, testing from a somatic tissue such as fibroblasts should be sent. Targeted panels designed for evaluation of somatic MDS mutations should not be used to evaluate germline mutations for a given gene, as the somatic mutation panels may be missing coverage at key regions of genes of interest or at mutation sites that differ in the acquired vs the germline setting. Because of the frequency of mutations that may be gene deletions or duplications, clinicians should also be sure that testing methods detect copy number variations.
Surveillance
Diagnosis of genetic predisposition to MDS provides an opportunity for prospective clinical surveillance and intervention before the development of advanced disease. AML carries an especially poor prognosis for many of these disorders, so early diagnosis offers an opportunity for timely bone marrow transplant before development of leukemia. The reader is referred to recent discussions of surveillance and management of patients with genetic predisposition to MDS.131,132
Treatment
Curative treatment of MDS for patients with germline MDS predisposition is a hematopoietic stem cell transplant. It is challenging that many genetic bone marrow failure syndromes are associated with excessive conditioning regimen-related toxicities and may require specialized reduced intensity conditioning regimens to minimize treatment-related toxicities that may, however, compromise ablation of the malignant clones.44,133-135 In addition, extrahematopoietic manifestations of germline predisposition disorders (Table 1) may further increase transplant risk. It is imperative to ensure that a related stem cell donor does not have the same genetic MDS predisposition disorder. Close monitoring for late effects is important in many of these patients, given the unique toxicities particular to their syndrome.136
Clonal evolution in germline MDS syndromes
Clonal evolution: cytogenetics
The genomic evolution of clonal expansion within germline MDS syndromes is an area of intense clinical and scientific interest. These disorders allow study of clonal evolution before the development of MDS, enabling researchers to investigate somatic events prospectively as they arise. This provides a unique opportunity to gain insights into the molecular events driving MDS and a clinical opportunity to potentially identify prognostic mutations, intervention strategies, and targeted therapies.
Cytogenetically abnormal clones frequently arise in these genetic predisposition disorders. Some of these clones may wax and wane, and their clinical significance is unclear.131,137 Critical to the workup of pediatric and young adult patients who present with hypocellular bone marrows is evaluation of both cytogenetics and fluorescence in situ hybridization studies, as the failing marrow often yields few metaphases, many of which may arise from lymphocytes rather than the marrow.131 This differs from aging-related sporadic MDS in which the marrow is often hypercellular and cytogenetics studies are often considered sufficient.
Clonal cytogenetics in germline MDS have provided unique insights into potential drivers of clonality that may be adaptive or maladaptive (Figure 2). Adaptive mutations provide a fitness advantage by complementing the intrinsic hematopoietic defect, whereas maladaptive mutations promote malignant transformation.
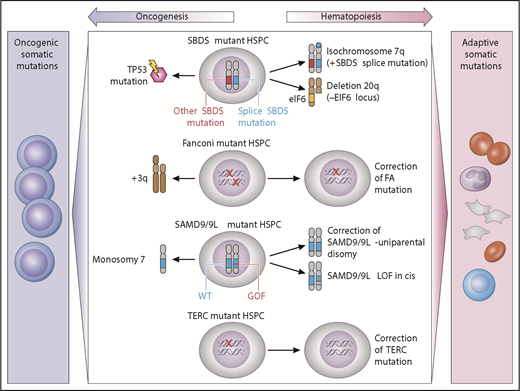
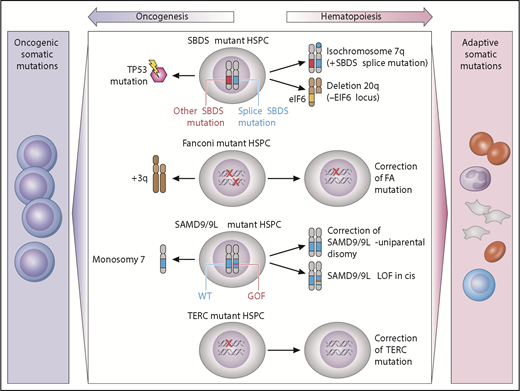
Model of germline genetic context in clonal evolution. Acquired somatic clonal mutations may be adaptive (improve hematopoiesis) or maladaptive (lead to transformation), depending on the underlying genetic MDS predisposition. In SBDS-mutant hematopoietic stem/progenitor cells, iso7q or del20q are common adaptive clonal aberrations, whereas TP53 somatic mutation is maladaptive. Fanconi anemia development of +3q is associated with progression to malignancy, whereas somatic reversion is not. In SAMD9/9L disorders, correction of the mutation by reversion or loss of mutant via truncation is adaptive, whereas development of monosomy is associated with MDS or AML. TERC-mutant HSPCs may undergo somatic reversion to regain 2 wild-type TERC alleles. GOF, gain of function; LOF, loss of function; HSPC, hematopoietic stem/progenitor cells; WT, wild type. Professional illustration by Patrick Lane, ScEYEnce Studios.
Model of germline genetic context in clonal evolution. Acquired somatic clonal mutations may be adaptive (improve hematopoiesis) or maladaptive (lead to transformation), depending on the underlying genetic MDS predisposition. In SBDS-mutant hematopoietic stem/progenitor cells, iso7q or del20q are common adaptive clonal aberrations, whereas TP53 somatic mutation is maladaptive. Fanconi anemia development of +3q is associated with progression to malignancy, whereas somatic reversion is not. In SAMD9/9L disorders, correction of the mutation by reversion or loss of mutant via truncation is adaptive, whereas development of monosomy is associated with MDS or AML. TERC-mutant HSPCs may undergo somatic reversion to regain 2 wild-type TERC alleles. GOF, gain of function; LOF, loss of function; HSPC, hematopoietic stem/progenitor cells; WT, wild type. Professional illustration by Patrick Lane, ScEYEnce Studios.
These adaptive clones are illustrated by 2 different alterations in SDS: isochromosome 7q and del20q. The iso7q region of duplication has been observed to duplicate the hypomorphic SBDS allele, which is predicted to potentially increase SDS protein and has not been associated with increased leukemia risk.138 Del20q clones also frequently arise in SDS. The deleted region spans EIF6, resulting in haploinsufficiency, and may provide a clonal fitness advantage by reducing translational stress.139
Adaptive somatic clones have also been described in patients with germline mutations in SAMD9/SAMD9L which reside on chromosome 7 (reviewed in Wlodarski et al21 and Davidsson et al24 ). Germline SAMD9 or SAMD9L gain-of-function mutations cause growth suppression of hematopoietic cells and resulting marrow failure.28,31 Patients with germline SAMD9 or SAMD9L mutations have a propensity to develop clones that have lost or inactivated the mutant SAMD9/SAMD9L allele. The mechanisms of this adaptation can be through truncating or loss of function mutations in cis with the mutant SAMD9 or SAMD9L allele, or through genetic reversion through duplication of the wild-type allele. Improvement in blood counts has been observed to accompany this somatic inactivation of the mutant SAMD9/SAMD9L allele. A second strategy to eliminate the mutant gene is to delete all or part of chromosome 7 carrying the mutant SAMD9/SAMD9L allele. Although this is predicted to result in improved cell growth, this comes at the cost of promoting development of monosomy 7 MDS.
The somatic reversion of FA genes also results in disease-context-specific adaptive clones. Somatic reversion occurs through point mutation, gene conversion, or recombination resulting in outgrowth of cells that have restored a wild-type FA allele.140,141 Somatic reversion is clinically important because it can lead to false-negative chromosomal breakage testing of lymphocytes. Mechanistically, the reversion event corrects the DNA repair phenotype and confers a growth advantage over the remaining germline mutant cells, but does not seem to drive myeloid malignancy. However, malignant clones may still arise from the remaining uncorrected marrow cells.140 In addition, somatic reversion via mitotic recombination has also been documented in several cases of DC with TERC mutation.142
Clonal evolution: somatic genomics
The availability of deep genomic sequencing allows previously unprecedented detailed and comprehensive analysis of the somatic mutational landscape in MDS. Patterns of somatic mutations differ between genetic MDS predisposition disorders, suggesting that germline genetic context may drive different mechanisms of clonal hematopoiesis.143 Recent studies have reported clonal hematopoiesis in young patients with germline GATA2, RUNX1, and SBDS mutations and SCN.144-146 Recent studies examining the molecular mechanisms underlying reduced penetrance in GATA2-mutated MDS and AML may involve epigenetic changes.147 Somatic mutations, similar to cytogenetic changes, may be adaptive or oncogenic. For example, patients with SCN frequently acquire mutations in the G-CSF receptor, CSF3R.113,145,148,149 CSF3R mutations were detected both in the neutropenic and AML phase of the disease. The majority of mutations caused truncations of the cytoplasmic domain of the CSF3R receptor, resulting in cytokine-independent signaling.150,151 Although CSF3R clonal mutations are prevalent in AML in patients with SCN, they have also been observed for many years in patients who have not transformed to AML, indicating that such mutations alone are not sufficient to cause AML.148,152
In contrast to SCN, clonal somatic mutations in SDS commonly involve TP53 and, when present in MDS, are associated with poor prognosis.4,145 Functional studies are needed to determine whether these mutations may relieve the translational stress of the ribosomal abnormality or provide a growth advantage. Longitudinal studies of TP53 mutations in SDS are lacking, so the prognostic significance of TP53 mutation arising outside the context of MDS is currently unclear.
Data are lacking to inform the clinical implications of somatic clonal abnormalities in germline MDS syndromes. Somatic clonal mutations should be considered within the context of the blood counts, marrow morphology, and clonal dynamics. Longitudinal studies integrating somatic alterations of serial samples with clinical outcomes are needed to understand the prognostic significance of somatic clonal abnormalities and potential targeted therapies toward premalignant clones.
Future directions
As with any rare disease, collaborative studies, longitudinal disease registries and clinically annotated sample repositories are essential to advance our understanding with the goal of improving clinical outcomes. Disease penetrance and cumulative cancer risk remain to be determined for many of these disorders. Major challenges include the elucidation of additional genes causing MDS, identification of prognostic biomarkers for risk stratification to inform surveillance and timing of HSCT, and development of personalized treatments for patients with genetic predisposition to MDS.
Acknowledgments
The authors apologize to all those whose work could not be cited because of space limitations. Recent reviews were cited in each section, and readers are referred to primary references therein. The authors are grateful to Mark Fleming and Suneet Agarwal for their contribution of images for figures.
The authors gratefully acknowledge support from the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (R24DK099808) (A.S.).
Authorship
Contribution: All authors provided conception and design, manuscript writing, and final approval of manuscript, and all authors are accountable for all aspects of the work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Akiko Shimamura, Karp 8210, 300 Longwood Ave, Boston, MA 08115; e-mail: akiko.shimamura@childrens.harvard.edu.