Abstract
Cells and the molecular processes underlying their behavior are highly dynamic. Understanding these dynamic biological processes requires noninvasive continuous quantitative single-cell observations, instead of population-based average or single-cell snapshot analysis. Ideally, single-cell dynamics are measured long-term in vivo; however, despite progress in recent years, technical limitations still prevent such studies. On the other hand, in vitro studies have proven to be useful for answering long-standing questions. Although technically still demanding, long-term single-cell imaging and tracking in vitro have become valuable tools to elucidate dynamic molecular processes and mechanisms, especially in rare and heterogeneous populations. Here, we review how continuous quantitative single-cell imaging of hematopoietic cells has been used to solve decades-long controversies. Because aberrant cell fate decisions are at the heart of tissue degeneration and disease, we argue that studying their molecular dynamics using quantitative single-cell imaging will also improve our understanding of these processes and lead to new strategies for therapies.
Introduction
Cellular behaviors are dynamic and different cell fates are acquired with changing needs of the organism, tissue, or cell population (Figure 1A). Understanding the molecular dynamics of cell fate decisions is important to understand and correct aberrant cellular behaviors during disease. Therefore, developing novel tools to unravel the mysteries of cell fate control is highly relevant for regenerative therapy.
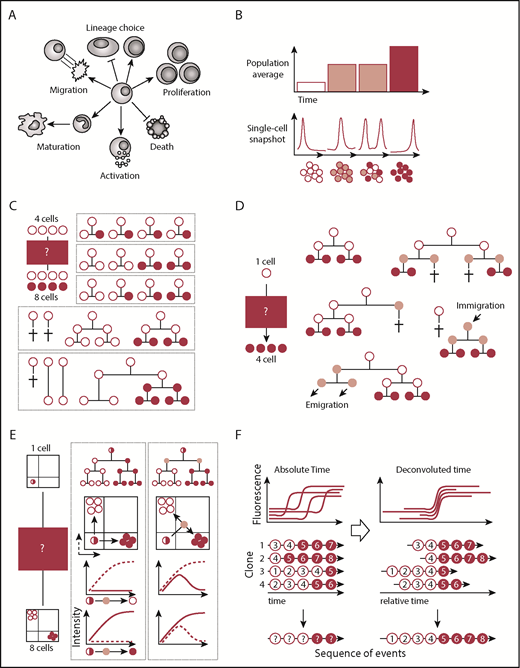
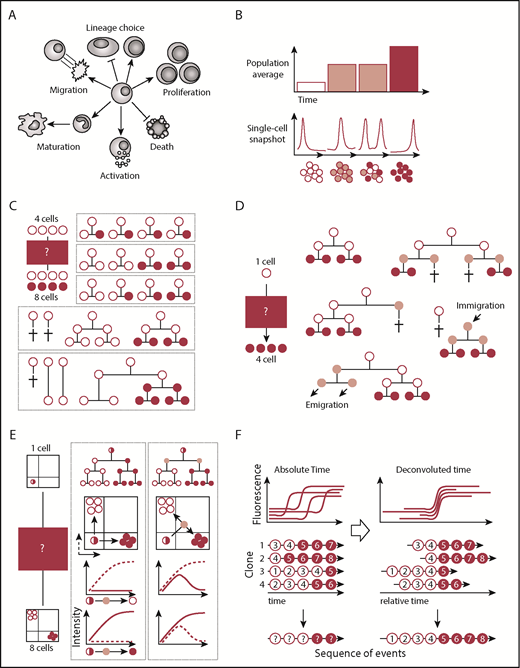
Population and/or single-cell snapshot data is nonambiguous and can yield many conclusions. (A) Cells can acquire multiple cell fates. (B) Single-cell snapshot analysis can reveal subpopulations otherwise masked in population averages. (C) Snapshot analysis is ambiguous and allows for alternative interpretations of the same data set. In this simple example, 4 input cells (white) give rise to 4 white cells and 4 red cells. Many more possibilities exist but are not displayed. (D) Alternative explanations also remain in clonal assays started from single cells. Here, a single cell input cell (white) generates 4 cells (red). Only some of many more possible alternative explanations are displayed. (E) Continuous quantification of fluorescence signals can reveal transient cellular states that would be missed by snapshot analysis. Here, a single input cell (white/red) gives rise to 4 white cells and 4 red cells expressing mutually exclusive lineage-promoting transcription factors. Differentiation into either cell type can occur directly or through a transient double-positive population. (F) Temporal deconvolution of continuously observed single-cell dynamics enables the undistorted analysis of cells at different developmental stages or cell cycle phases. Temporally aligned single-cell dynamics can reveal the sequence of molecular events prior to and after differentiation (indicated by red numbers).
Population and/or single-cell snapshot data is nonambiguous and can yield many conclusions. (A) Cells can acquire multiple cell fates. (B) Single-cell snapshot analysis can reveal subpopulations otherwise masked in population averages. (C) Snapshot analysis is ambiguous and allows for alternative interpretations of the same data set. In this simple example, 4 input cells (white) give rise to 4 white cells and 4 red cells. Many more possibilities exist but are not displayed. (D) Alternative explanations also remain in clonal assays started from single cells. Here, a single cell input cell (white) generates 4 cells (red). Only some of many more possible alternative explanations are displayed. (E) Continuous quantification of fluorescence signals can reveal transient cellular states that would be missed by snapshot analysis. Here, a single input cell (white/red) gives rise to 4 white cells and 4 red cells expressing mutually exclusive lineage-promoting transcription factors. Differentiation into either cell type can occur directly or through a transient double-positive population. (F) Temporal deconvolution of continuously observed single-cell dynamics enables the undistorted analysis of cells at different developmental stages or cell cycle phases. Temporally aligned single-cell dynamics can reveal the sequence of molecular events prior to and after differentiation (indicated by red numbers).
Cell fate decisions have classically been studied at discrete time points as population averages and at the single-cell level (Figure 1B). Multiple high-throughput single-cell approaches like multiplexed quantitative polymerase chain reaction,1 fluorescence in situ hybridization,2 RNA sequencing,3 mass cytometry,4 and immunostainings5 are available today and enable the simultaneous analysis of thousands of cells and genes at the RNA and/or protein level. These approaches have revealed cellular heterogeneity of cell populations previously thought to be homogenous.6,7 Novel subpopulations, single-cell trajectories, and transition states were inferred using computational approaches like pseudotemporal ordering,3,8 iterative clustering and guide gene selection,9 or Boolean models.1,10 The actual timing and order of cell fate decisions are not measured with these approaches but are reconstructed assuming that differentiation is a continuous unidirectional process with constant speed. Molecular oscillations and discontinuous cell fate transitions accomplished by, for example, asymmetric cell divisions (ACDs) are not considered. Therefore, snapshot data, even from single cells, cannot completely elucidate the exact timing and order of events underlying cell fate decisions. As depicted in Figures 1 and 2, snapshot data typically allow multiple alternative interpretations of the same data set, even with few starting cells.11,12 In most experiments, many thousands of cells of different subpopulations are intermingled and cannot be purified prospectively. Thus, data obtained from cell populations will be averaged over multiple subpopulations and developmental stages, thus losing rare subpopulations. Single-cell snapshot approaches ameliorate this issue, but it remains unclear whether differences between individual clones reflect different cell types, different developmental stages, or cell cycle phases of the same cell type over time.
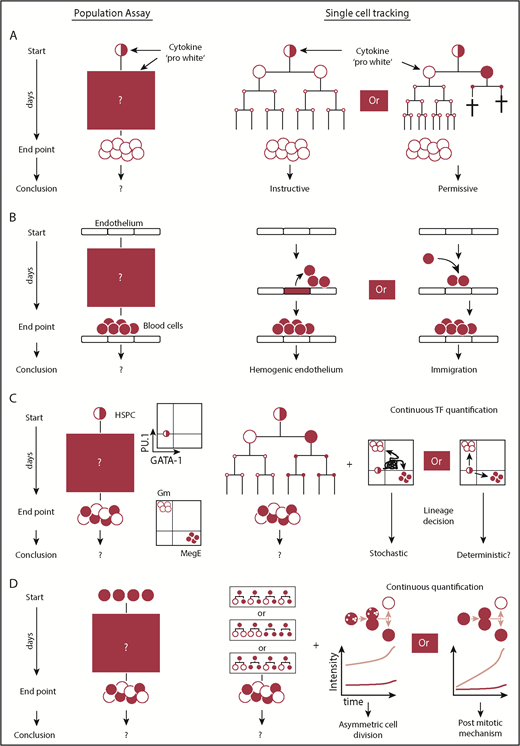
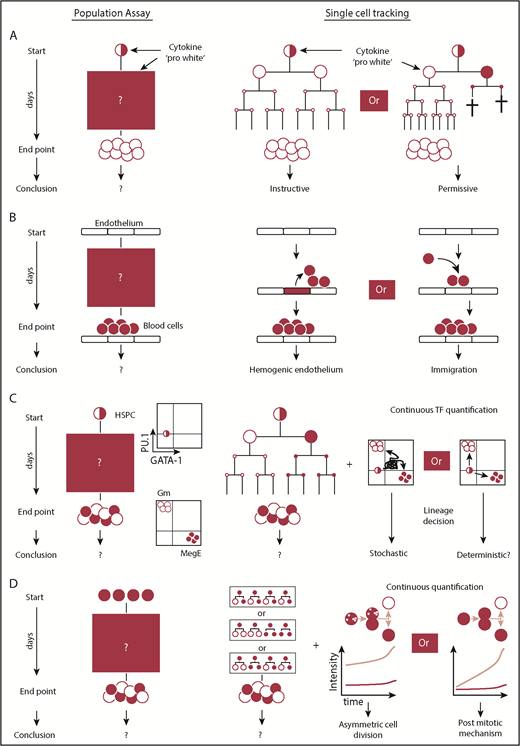
Questions that require continuous single-cell observations to untangle alternative interpretations. (A) Snapshot analysis is not able to discern between instructive and permissive roles of cytokines during lineage decision. The selective model requires cell deaths prior to terminal differentiation. Proof for the absence of cell death requires knowledge of the fates of every single cell over time. Using this approach, it was demonstrated that certain cytokines can instruct lineage choice. (B) The presence of blood cells on top of endothelium can be explained by generation of blood cells from hemogenic endothelium or from alternative sources. The direct observation of endothelial to hematopoietic transition using continuous observation provided direct evidence for the existence of hemogenic endothelium. (C) Lineage decisions based on the stochastic fluctuations of 2 antagonistic lineage promoting transcription factors require a (transient) double-positive cell state. By demonstrating the absence of the PU.1/GATA-1 double-positive cell state during most differentiations into the myeloid or megakaryocytic-erythroid lineage, this model could be refuted as the central mechanism of myeloid lineage choice. (D) The acquisition of different daughter cell fates can be regulated by mechanisms acting after or during cell division. Untangling both mechanisms requires continuous quantitative single-cell observation and the demonstration that different daughter cell fates can be predicted by the asymmetric inheritance of factors during mitosis.
Questions that require continuous single-cell observations to untangle alternative interpretations. (A) Snapshot analysis is not able to discern between instructive and permissive roles of cytokines during lineage decision. The selective model requires cell deaths prior to terminal differentiation. Proof for the absence of cell death requires knowledge of the fates of every single cell over time. Using this approach, it was demonstrated that certain cytokines can instruct lineage choice. (B) The presence of blood cells on top of endothelium can be explained by generation of blood cells from hemogenic endothelium or from alternative sources. The direct observation of endothelial to hematopoietic transition using continuous observation provided direct evidence for the existence of hemogenic endothelium. (C) Lineage decisions based on the stochastic fluctuations of 2 antagonistic lineage promoting transcription factors require a (transient) double-positive cell state. By demonstrating the absence of the PU.1/GATA-1 double-positive cell state during most differentiations into the myeloid or megakaryocytic-erythroid lineage, this model could be refuted as the central mechanism of myeloid lineage choice. (D) The acquisition of different daughter cell fates can be regulated by mechanisms acting after or during cell division. Untangling both mechanisms requires continuous quantitative single-cell observation and the demonstration that different daughter cell fates can be predicted by the asymmetric inheritance of factors during mitosis.
Time-lapse imaging can overcome these obstacles and, in many ways, it is the ultimate approach to understand dynamic single-cell behavior. Cell fates, like migration, proliferation, cell death, and differentiation, are quantified with absolute accuracy. Because all changes in cell behavior and marker expression are recorded for every single cell over time, noise introduced by, for example, heterogeneous developmental stages and lack of synchronization in a population can be corrected by temporal deconvolution (Figure 1F).
The information stored in millions of images enables the study of correlations and causalities at the single-cell level with unprecedented detail. However, the analysis of dynamic changes in a multitude of fluorescent and morphometric parameters is complex and requires interdisciplinary know-how in biology, computer programming, and digital imaging. Because most biologists did not receive this training, few expert laboratories have managed to establish this demanding technology successfully. However, overcoming these challenges offers novel ways to quantitatively understand biological processes.
Continuous long-term single-cell quantification is important for understanding dynamic processes, and it has contributed answers for many tissues with clear 3-dimensional organization.13,14 It is even more important for the hematopoietic system, in which the lack of firm adhesion in a tissue, constant migration and mixing, and the incompletely understood 3-dimensional organization make it difficult to identify specific cell types and maturation stages, differentiation trajectories, relevant niches, and, thus, molecular mechanisms underlying health and disease. We will focus mostly on time-lapse studies with hematopoietic cells for the rest of this review.
Early time-lapse studies
The origins of time-lapse microscopy can be traced back to Etienne-Jules Marey,15,16 who assembled the first documented time-lapse microscope in 1891 and filmed red blood cells.15,17 In 1895, the Lumiére brothers patented the cinematograph, a device capable of “manipulating” time by speeding up or slowing down recordings during playback.18 Shortly thereafter, cinematographers started to look at microscopic specimens. In 1903, Duncan recorded, in one of the earliest microcinematographic movies of living organisms, the movement of cheese mites.15 In 1907, Ries filmed sea urchin development,19 and, in 1909, Comandon demonstrated that disease-causing and nondisease-causing syphilis bacteria could be distinguished based on their motility.16
After the development of tissue culture in 1907 by Harrison20 and the first commercial microcinematograph in 1914,21 microcinematography was adopted by Carrel and Rosenberger to study the movement of fibroblasts and macrophages in culture.22 In one of the earliest methods articles about time-lapse experiments, they advocated cinematography to researchers “who have not yet realized the great possibilities of the motion-picture camera in research laboratories.”23 Canti and Lewis were the first to use time-lapse microcinematography to study the developing embryo.15,24 Canti’s movies are particularly impressive because he managed to record the growth of an entire chicken thigh bone for 2 weeks. Maintaining the viability of these cultures for such extended periods of time while keeping the specimen in focus and the microscope running is not trivial, even today.
Despite these successes, early microcinematographic research had many critics. The Nobelist Medawar stated that cell biologists have been “delighted, distracted, and beguiled by the sheer beauty” of cells on film, and, as a result, have missed the opportunity to use cytological methods to “solve biological problems.”21 This impression was shared by many cytologist at the time and only slowly began to change after Abercrombie demonstrated that time-lapse imaging could be used to extract quantitative information about the cellular movements of fibroblasts.15,25 Irene Boll, a pioneer in time-lapse cinematography of hematopoietic cells, quantified their maturation, locomotion, mitosis, and apoptosis.26-29 However, Boll’s work remained an exception; it took until the 1990s, with the arrival of cheap electronics and solid-state cameras enabling the acquisition and storage of digital images, for time-lapse imaging to become more widespread.
Technical and organizational considerations
Time-lapse studies require meticulous preparation, and many unexpected biological, technical, and organizational caveats exist.30 For instance, institutional information technology departments are often not prepared to deal with the vast amounts of data acquired by modern microscopes (terabytes per day). There is no single best way to conduct time-lapse experiments. Depending on the biological question, optimal settings need to be determined iteratively. One often underestimated issue is cell survival, which is commonly impaired by phototoxicity, medium evaporation, and pH changes. The acquisition of aesthetic images often requires experimental settings exceeding what cells can tolerate. All approaches should always be adjusted to minimize phototoxicity by lowering imaging frequencies and light intensities. On top of that, culture conditions have to be chosen carefully to enable “normal” cell behavior as much as possible in vitro.
Once the first terabytes of data are acquired, the real challenge begins: data storage, processing, curation, and analysis. Terabytes to petabytes of data scattered across millions of files require automated solutions; however, commercial ready-to-use software is not available or is too error prone, and manual analysis often remains the only way to extract data reliably. Many existing image analysis tools like ImageJ/Fiji,31,32 Icy,33 Cell Profiler,34 and LEVER35 provided partial support for single-cell tracking but remain inefficient to use. Solutions like tTt,36,37 specifically developed for long-term single-cell tracking, have only recently become available. These tools fill a crucial gap, but additional customized solutions are still required and need to be integrated into a greater data analysis pipeline addressing file/data conversion, background correction, segmentation, quantification, tracking, data curation, and analysis. A central, and often underestimated, aspect is the efficient implementation of these individual parts and the way in which they are tethered. A single suboptimal or unreliable step of this pipeline can easily delay data analysis by weeks to months.
The challenge to image hematopoietic cells
While the early microcinematographic movies were only a few hours long,38,39 in the 21st century hematologists started to use time-lapse imaging to track single cells for days to weeks in vitro. Owing to their high motility, tracking hematopoietic cells long-term proved to be challenging. Because the size of tissue culture plates usually exceeds the camera field of view by several fold, it is impossible to cover the entire well and cells quickly leave the field of view. The introduction of motorized microscopy stages made the acquisition of several adjacent fields of view possible, and following cells across multiple positions became feasible. However, owing to the high cell motility, images still needed to be taken every few minutes. Only few adjacent positions could be covered before cells would have moved too far to assign their identity unequivocally. After the introduction of methylcellulose had revolutionized hematology by restricting cell motility and enabling clonal in vitro assays,40 hematologists were again looking for ways to keep track of clones and cells over time. This time it had to be compatible with the many requirements for time-lapse imaging.
Dykstra et al addressed this problem by scraping micro wells into a silicone gel casted onto a coverslip. This allowed imaging of multiple physically separated hematopoietic stem cells (HSCs) simultaneously in a single field of view. Using custom-made tracking software, they annotated cell morphology, location, and cellular kinships over time and retrieved and transplanted individual clones into mice. By correlating the genealogical history and annotated morphologies with the ability of in vitro cultured clones to reconstitute mice, they demonstrated that HSC activity in vitro is associated with longer cell cycle times and the absence of uropodia.41
Lutolf et al42 engineered hydrogel-based microwell arrays to restrict cell movement and studied the effects of secreted and tethered factors on HSCs. They imaged HSCs in various culture conditions, retrieved and transplanted individual clones, and discovered that the Wnt3a and N-cadherin–mediated reduction of HSC proliferation and asynchronous division times correlate with HSC function42 and that lipid raft clusters can be used to infer HSC activation.43
Hawkins et al44 cultured B cells in wells that are small enough to image their entire content with a single camera field of view. Using custom tracking software, they tracked B cells and described that division times between siblings strongly correlate and that the proliferative potential of B-cell clones is related to the founder cell size prior to its first division. Based on this observation, they speculated that the simple dilution of factors during division might control the cells’ proliferative potential and proposed an internal cellular mechanism. Later, 2 heritable factors could be shown to be sufficient to explain the observed familial correlations of cell division and death,45 and different cell fate decisions are pursued autonomously by intracellular stochastic competition,46 whereas autonomous programs in CD8+ T cells can be extended by multiple inputs that sum linearly to control the strength of the T-cell response.47
Lecault et al restricted cell movement in a microfluidic platform with thousands of chambers to culture and image nonadherent cells.48 Using a microfluidic chip capable of automatically exchanging medium and retrieving clones, they studied the impact of temporally varied stimulation of 2 stem cell factor (SCF) concentrations on HSC potential. In contrast to high SCF concentrations, low SCF had been demonstrated to lead to a loss of HSC function in vitro. Culturing HSCs in low SCF for 8, 16, 24, or 48 hours, followed by high SCF, Lecault et al asked whether the effects of low SCF exposure are reversible and, if so, for how long. Prolonged exposure to low SCF led to decreased viability between 16 and 24 hours; after that, cells could not be rescued when exposed to high SCF. Sekulovic et al characterized the effects of nucleoporin 98–homeobox A10 homeodomain (NUP98-HOXA10hd) expression on HSCs.49 Upon expression and in vitro culture in hematopoietic stem and progenitor cells (HSPCs), NUP98-HOXA10hd had been shown to facilitate a >1000-fold expansion of cells with stem cell properties. Whether this effect was mediated by modulating cell survival, proliferation, or differentiation was unclear. Although the exact mode of action was not identified, altered proliferation of NUP98-HOXA10hd–transfected cells could be excluded as the underlying mechanism of expansion.
These studies exemplify that microfluidics is more than tiny tissue culture chambers. It offers novel ways to dynamically manipulate cells at defined time intervals. The capability to exchange medium at defined flow rates is particularly important for nonadherent hematopoietic cells because these cells are easily flushed away.50
The combination of motorized stages and novel custom-written tracking software, including user interface, enabled tracking across multiple fields of view without cell loss. In contrast to previous studies, multiple cell fates, like adhesion, morphology, division, death, and fluorescent marker expression (on/off), over many generations were annotated and used to create cellular genealogical trees,51,52 and a recent extension now allows the quantification of fluorescence reporter intensities.7,53,54
These studies were only possible because of custom hardware and/or software solutions in specialized laboratories, and their complexity still prevents the widespread application of time-lapse imaging. The high motility of hematopoietic cells remains the major problem for longitudinal single-cell tracking. Although cell motility can be restricted by microstructures, extracellular matrix coatings, or stroma cocultures, these solutions have their own drawbacks.55-57 Simple coating with anti-CD43 or anti-CD44 has recently been described to drastically reduce hematopoietic cell motility in vitro without affecting cell behavior.58 On this coating, cells do not leave the field of view anymore and form 2-dimensional colonies. This enables the observation of individual clones without the need for microengineered structures to restrict cell movement; more importantly, it increases the throughput of time-lapse experiments as a result of the much-reduced requirements in imaging frequency while greatly simplifying cell tracking.
Hemogenic endothelium: origins of hematopoietic cells in the embryo
The origins of the first hematopoietic cells in the mammalian embryo were disputed for decades.59 Different cellular origins, including hemogenic endothelium, had been proposed, but definitive proof was lacking, because the generation of blood cells could not be observed directly at the single-cell level (Figure 2B). Eilken et al51 addressed this issue by using continuous long-term single-cell imaging of mouse mesodermal cells. These cells and their progeny were followed for up to 1 week throughout the generation of blood cells, and cell morphology and molecular expression of endothelial and bloods cells were monitored using multiple fluorescence markers.60 The direct observation that endothelial cells emigrate from colonies of functional endothelial cells with tight junctions, transform morphologically into suspension cells, and upregulate hematopoietic marker molecules provided direct evidence for the existence of hemogenic endothelial cells.51
The instructive role of cytokines during lineage decision
Cytokines regulate cellular behavior and influence cell fate decisions by activating a multitude of intracellular signaling pathways. They control lineage choice and regulate the number of different mature cell types. However, the way in which cytokines execute their function remained disputed for decades.61 Cytokines could instruct lineage choice in multipotent progenitors or only select for already lineage-restricted cells by promoting their survival or proliferation (Figure 2A). In the latter case, lineage choice would be determined independently of cytokines. Because the instructive and permissive models lead to the same lineage and culture outcome, this question could not be addressed using snapshot analyses. Individual cell fate decisions in both models differ over time, and the occurrence of cell death would be expected if the cytokines act in a permissive manner. In contrast, an instructive mechanism could only be proven if even single-cell deaths throughout differentiation could be excluded. Therefore, the prerequisite to untangle these 2 models was to know each individual cell fate until differentiation with absolute accuracy, and it required continuous single-cell observation. The exclusive differentiation of granulocyte-macrophage progenitors (GMPs) into granulocytes or macrophages in the presence of granulocyte colony-stimulating factor (G-CSF) or macrophage colony-stimulating factor (M-CSF), respectively, was used to answer this question. By demonstrating that the differentiation exclusively into a single lineage occurs in the absence of cell death, a solely selective process could be excluded, and direct evidence of an instructive function of cytokines was provided for G-CSF and M-CSF in GMPs.52
M-CSF signaling is activated upon binding to its receptor. Multiple tyrosine residues of its cytoplasmic domain get phosphorylated and bound by adaptor proteins that initiate downstream signaling. Individual tyrosine residues had previously been suggested to activate specific signaling pathways, but contradictory conclusions were drawn from snapshot analysis using cell lines. Therefore, the role of individual pathways underlying the instructive role of M-CSF regulating cell fate decisions remained unclear. Combined loss- and gain-of-function experiments with single-cell tracking demonstrated that Src signaling is sufficient for M-CSF–instructed differentiation of primary GMPs into macrophages.57
Mechanism of myeloid lineage decisions: the PU.1/GATA-1 switch paradigm
The prevailing model for early myeloid lineage decisions assumed cell-intrinsic stochastic fluctuations of the lineage-associated transcription factors PU.1 and GATA-1. Earlier reports had shown that both factors possess the capacity for lineage reprogramming upon overexpression and the ability to positively autoregulate and cross-inhibit their expression,62 as well as that their expression dynamics matter.63 Snapshot RNA expression data suggested that early hematopoietic progenitors express both transcription factors.64 Using a novel reporter mouse, no coexpression of these factors was found at the protein level,55 but snapshot data could not exclude the possibility that, upon differentiation, cells could quickly pass through a transient state in which both factors are expressed (Figure 2C). Continuous single-cell quantification of PU.1 and GATA-1 protein levels throughout the entire differentiation process finally provided proof that the majority of differentiating HSPCs do not, in fact, pass through a PU.1/GATA-1 double-positive state before lineage decision making and that those that do, always differentiate into a GATA-1–expressing end state. This demonstrated the prevailing stochastic model to be incorrect and that these transcription factors only execute, rather than decide, myeloid lineage fate.65,66
Identifying novel culture conditions for HSC maintenance in vitro
HSCs have been successfully used to repair patient’s bone marrow for decades. However, their scarcity restricts their clinical application. Long-term in vitro expansion of HSC numbers could overcome this limitation; however, despite decades-long research, attempts to identify robust expansion culture conditions have failed. Reasons for loss of HSC potential in vitro remained unclear. In one study, HSCs could be maintained for several weeks in vitro when cocultured with AFT024 stroma cells.67 The molecular players remained unknown, because differential gene-expression analysis between supportive and nonsupportive stroma revealed >1000 putative candidate genes. Given this number of candidates, the identification of relevant factors using lengthy functional readouts was impossible and not pursued. Characterizing the differential cellular behaviors of HSCs in supportive and nonsupportive stroma cocultures over time revealed that supportive stroma mediated HSC survival. Time-lapse imaging showed that the prosurvival factor(s) must be contact dependent and enabled to narrow down putative candidates. Knocking down candidates in supportive AFT024 stroma cells demonstrated that the prosurvival effect is dermatopontin dependent.
The quest for ACD in HSCs
Based on observations in invertebrate model systems, it had been speculated that hematopoietic lineage bifurcations could be controlled by ACD. During ACD, different future daughter cell fates are deterministically instructed by a mitotic mechanism. Importantly, symmetric and asymmetric cell fates can be accomplished postmitotically or by mechanisms related to division. Therefore, a prerequisite to demonstrate the existence of ACD is to untangle these 2 scenarios (Figure 2D) and requires continuous single-cell observation (eg, by demonstrating that the asymmetric inheritance of factors during division predicts future daughter cell fates).
ACD of lymphocytes had originally been suggested by the coinheritance of factors in fixed samples.68 Later, time-lapse imaging demonstrated the asymmetric inheritance of factors in CD4+ T cells,69 CD8+ T cells,70,71 B cells,72 and DN3a thymocytes.73 However, the relevance of lymphocyte ACD in vivo remains disputed.
Cell polarity74 and asymmetric inheritance of molecules during HSPC division have been suggested,75-77 but a correlation with daughter cell fates could not be shown.78 Therefore, the functional relevance of this asymmetric inheritance and, thus, the existence of ACD in HSCs, could not be proven, and it remains possible that the observed asymmetries only reflect imprecise distribution of irrelevant factors. More convincing evidence for hematopoietic ACD comes from human multipotent progenitors (MPPs). A combination of imaging and daughter cell separation suggests that the asymmetric inheritance of CD133 correlates with lineage potential.79 However, the number of analyzed paired daughter cells is low, and the inheritance of CD133 was not quantified. Therefore, it remains to be seen whether these results can be verified by other investigators.
Integrating dynamic molecular quantification with high-dimensional omics snapshot data
Despite the advantages of time-lapse imaging, the number of extractable parameters over time is low compared with high-throughput snapshot analysis like single-cell RNA sequencing. Combining both approaches would offer additional insights when individual cells are isolated from time-lapse experiments and disruptively analyzed. A prerequisite for this analysis is to unequivocally keep track of cells during isolation.80 However, computational tools required to integrate and analyze cellular kinship, quantitative time-series, and high-dimensional omics data are not available, and retrieving relevant information still requires hardware and software development.
Tracking hematopoietic cells in vivo
Despite its recent success, in vitro time-lapse imaging is done in an artificial environment. This is often important in its own right, because cell expansion or manipulation and characterization for therapeutic purposes will usually be done in vitro. In this case, understanding cell behavior and molecular control is important, even if they differ from the in vivo situation. However, it is important to keep in mind that the cellular and molecular behaviors measured with cultured cells might differ from those in vivo. Therefore, it is crucial to select cell types, culture systems, and questions to be asked that allow relevant insights from in vitro studies. In addition, insights from comprehensive quantitative in vitro studies should be complemented with more focused longitudinal noninvasive in vivo single-cell tracking. Intravital microscopy has been used in worm,81 fly, and zebrafish.82 However, in most mammalian tissues, technical limitations still usually prevent the long-term observation of single cells. Magnetic resonance imaging, positron emission tomography, single-photon emission computed tomography, fluorescence imaging, and bioluminescence imaging (see Nguyen et al83 for additional details) have been used for in vivo imaging but are restricted by constraints that living organisms can only be immobilized for short periods. Analogous to in vitro single-cell tracking, the temporal resolution required to unequivocally track single cells over time is a major challenge. Despite these time limitations, especially in situations in which cell fate decisions can take many days, short-term observations have provided insights into migration, homing, and engraftment of HSCs and leukemic cells, their localization,84-89 and the generation of HSCs from the ventral wall of the aorta in zebrafish.90
In contrast to highly motile hematopoietic cells, cells of solid tissues migrate slowly. Therefore, image acquisition every few hours is sufficient to assign single-cell identities reliably when spermatogonial progenitor cells in mouse testis91 and adult-born neurons were tracked for multiple days.92 However, these studies are not applicable to hematopoietic cells, whose longitudinal study in vivo requires higher temporal resolution but which might finally become feasible when single-cell detection in freely moving animals comes of age.93
Recently, cellular barcoding has emerged as an important approach to reconstruct cellular phylogenies in vivo.94 Although cellular and molecular dynamics cannot be inferred using this approach, the clonal evolution and output over many decades can be recapitulated, much extending the “observable” time frames beyond what is technically feasible by imaging.
Future perspective
Understanding dynamic cellular and molecular behaviors over time requires continuous observation and single-cell quantification. Seminal time-lapse studies demonstrated how direct observations of cell fate decisions over time provide direct evidence instead of ambiguous conclusions based on indirect bulk and single-cell snapshot data. Because of that, single-cell tracking is being used increasingly.95-97 However, the analysis of cellular genealogies remains challenging, and most data sets remain only partially analyzed.97-99 An interdisciplinary effort is required to develop novel analysis tools and statistical methods, as well as to make them available to a broad range of researchers in easy-to-use interfaces. Their application will also require a new generation of quantitative biologists with interdisciplinary know-how about image analysis, computational methods, and statistics.
Acknowledgments
The authors acknowledge support by the Swiss National Science Foundation.
Authorship
Contribution: D.L. and T.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Timm Schroeder, Department of Biosystems Science and Engineering (D-BSSE), Eidgenössische Technische Hochschule (ETH) Zurich, Mattenstr 26, 4058 Basel, Switzerland; e-mail: timm.schroeder@bsse.ethz.ch.