Key Points
Using central review in a multi-institution study, 28.2% of submitted cGVHD cases in children were reclassified using the NIH-CC.
The NIH-CC are feasible and reliable for children, but further refinement of criteria, specifically for pediatric cGVHD, is needed.

Abstract
Chronic graft-versus-host disease (cGVHD) and late acute graft-versus-host disease (L-aGVHD) are understudied complications of allogeneic hematopoietic stem cell transplantation in children. The National Institutes of Health Consensus Criteria (NIH-CC) were designed to improve the diagnostic accuracy of cGVHD and to better classify graft-versus-host disease (GVHD) syndromes but have not been validated in patients <18 years of age. The objectives of this prospective multi-institution study were to determine: (1) whether the NIH-CC could be used to diagnose pediatric cGVHD and whether the criteria operationalize well in a multi-institution study; (2) the frequency of cGVHD and L-aGVHD in children using the NIH-CC; and (3) the clinical features and risk factors for cGVHD and L-aGVHD using the NIH-CC. Twenty-seven transplant centers enrolled 302 patients <18 years of age before conditioning and prospectively followed them for 1 year posttransplant for development of cGVHD. Centers justified their cGVHD diagnosis according to the NIH-CC using central review and a study adjudication committee. A total of 28.2% of reported cGVHD cases was reclassified, usually as L-aGVHD, following study committee review. Similar incidence of cGVHD and L-aGVHD was found (21% and 24.7%, respectively). The most common organs involved with diagnostic or distinctive manifestations of cGVHD in children include the mouth, skin, eyes, and lungs. Importantly, the 2014 NIH-CC for bronchiolitis obliterans syndrome perform poorly in children. Past acute GVHD and peripheral blood grafts are major risk factors for cGVHD and L-aGVHD, with recipients ≥12 years of age being at risk for cGVHD. Applying the NIH-CC in pediatrics is feasible and reliable; however, further refinement of the criteria specifically for children is needed.
Medscape Continuing Medical Education online
![]()
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.00 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine's (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider's responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 332.
Disclosures
Author Justin T. Wahlstrom was employed by Pharmacyclics, Inc. Associate Editor Robert Zeiser, CME questions author Laurie Barclay, freelance writer and reviewer, Medscape, LLC, and the remaining authors declare no relevant financial relationships.
Learning objectives
Upon completion of this activity, participants will be able to:
Describe use of the National Institutes of Health Consensus Conference (NIH-CC) criteria to diagnose pediatric chronic graft-versus-host disease (cGVHD), according to a prospective, multi-institution study
Determine the frequency of cGVHD, late acute graft-versus-host disease (L-aGVHD), and pulmonary cGVHD in children by using the NIH-CC criteria, according to a prospective, multi-institution study
Identify clinical features of and risk factors for cGVHD and L-aGVHD using the NIH-CC criteria, according to a prospective, multi-institution study
Release date: July 18, 2019; Expiration date: July 18, 2020
Introduction
Chronic graft-versus-host disease (cGVHD) is a major long-term complication of allogeneic hematopoietic stem cell transplantation (allo-HSCT), resulting in adverse physical and psychological functioning,1-3 disability,4 and poor quality of life.5,6 Further, cGVHD remains the leading cause of late nonrelapse mortality following allo-HSCT.7 The high incidence of cGVHD in adults, between 30% and 65%,8,9 has resulted in an extensive body of cGVHD literature in this population.
By comparison, cGVHD in infants, children, and adolescents has been relatively understudied, with most cGVHD studies not including pediatric patients.9 The reasons for this include fewer transplants being performed compared with adults, as well as a lower incidence of cGVHD in children, making cGVHD research in this age group challenging. This is particularly true for pediatric-specific prospective multi-institution cGVHD studies, which are lacking. The true incidence of cGVHD in children is imprecisely known, with estimates from registry studies suggesting 6% to 33%, depending upon graft source.10-12 For children with cGVHD, the disorder may become a major cause of morbidity and mortality.13-15 Given their longer life expectancy, studying cGVHD in children remains important to improve the outcome of pediatric allo-HSCT.
A major concern with cGVHD research revolves around the challenges with clinical cGVHD evaluation, resulting in inconsistencies in reporting and, therefore, quality of cGVHD data.16 The original Seattle cohort defining any graft-versus-host disease (GVHD) manifestation occurring after day +100 as being “chronic”17 is no longer accepted, given contemporary knowledge regarding the biology of acute GVHD (aGVHD) and cGVHD, which are now understood as related, but distinct, pathophysiologic entities with different clinical features.18,19 A major advancement in the field came with the publication of the 2005 National Institutes of Health Consensus Criteria (NIH-CC) for the diagnosis and staging of cGVHD, which were later updated in 2014.20,21 The NIH-CC defined cGVHD as requiring the presence of ≥1 diagnostic cGVHD sign or symptom, or 1 distinctive cGVHD manifestation with supporting laboratory or other organ system involvement. Further, the NIH-CC separated features that were common to both aGVHD and cGVHD as not being diagnostic for cGVHD, regardless of their time of development posttransplant. Finally, manifestations of aGVHD present after day +100 are now called late acute graft-versus-host disease (L-aGVHD) to differentiate them from cGVHD. Considerable effort to validate the NIH-CC in adults has occurred, with the 2014 NIH-CC considered the most comprehensive criteria for cGVHD assessment.16 However, clinical applicability of the NIH-CC in children has received little attention outside of 2 single-institution studies that retrospectively reclassified children with cGVHD according to the criteria.22,23 Indeed, the diagnostic criteria for cGVHD were developed primarily from adult data, and it remains unknown whether the NIH-CC can be easily applied to children given the potential difficulties with assessment in pediatrics (eg, young children may not be able to perform pulmonary function tests [PFTs] because of their age) and how little is known about L-aGVHD in this population.
The Applied Biomarkers of Late Effects of Childhood Cancer/Pediatric Blood and Marrow Transplant Consortium 1202 study (ABLE/PBMTC 1202) was a multi-institution prospective study aimed at discovering prognostic biomarkers for pediatric cGVHD.24 Integral to the validity of the biomarker studies was the proper classification of the GVHD status of enrolled subjects according to the 2005 NIH-CC, including near real-time review of cGVHD manifestations after diagnosis and central adjudication when required. This study presents results from these clinical GVHD assessments and documents the benefits and challenges with using the NIH-CC for cGVHD and L-aGVHD evaluation in pediatrics, within the context of a multi-institution study. Our objectives were to evaluate cGVHD and L-aGVHD diagnosis in children according to the NIH-CC and, in particular, to determine: (1) whether the NIH-CC, based primarily upon adult data, operationalize well in transplant recipients <18 years of age, (2) the frequency of cGVHD and L-aGVHD in pediatrics, and (3) the clinical presentations of, and risk factors for, both disorders. This study represents the largest cohort of prospectively collected children diagnosed with cGVHD and L-aGVHD according to the NIH-CC published to date.
Patients and methods
Study design
Twenty-seven pediatric transplant centers (20 in the United States, 6 in Canada, 1 in Europe) enrolled 302 patients between 8 August 2013 and 16 February 2017. All sites had institutional ethics board approval. Following written informed consent/assent, patients were enrolled before the start of conditioning and followed prospectively for 12 months (± 1 month) following hematopoietic stem cell transplantation (HSCT) for the development of L-aGVHD and cGVHD. Eligibility included age 0 to 18 years at the time of allo-HSCT, as well as any transplantable diagnosis, graft source, and GVHD prophylaxis regimen. Enrollment was initially restricted to myeloablative conditioning regimens25 and excluded haploidentical transplants; however, with changing transplant trends, the study was amended to include reduced-intensity conditioning and haploidentical transplantation. Exclusion criteria included second or greater allo-HSCT, ex vivo T-cell depletion, and inability of a patient to follow up at the transplant center for 1 year post-HSCT. Transplant centers evaluated patients according to their routine practices after HSCT, with scheduled assessments at day +100 (±14 days), 6 months (±1 month), and 12 months (±1 month). All subjects reported with cGVHD to the study by the local institution are included. Patients with nonengraftment (n = 14), patients with early relapse or toxic mortality before day +114 (n = 11 and n = 16, respectively), patients who were withdrawn for any reason (n = 14), and patients who did not proceed to transplant (n = 4) were excluded. Patients meeting these criteria were placed into 1 of 3 groups at the end of follow-up: control group (no L-aGVHD or cGVHD), L-aGVHD only, and cGVHD (including overlap syndrome).
Clinical data capture
Detailed case report forms (CRFs) were completed at 4 time points (preconditioning, day +100, and 6 and 12 months), reflecting standard transplant data before and after transplant (supplemental Data, Main Case Report Forms; available on the Blood Web site). Classical aGVHD was defined as aGVHD manifestations (maculopapular rash, nausea/vomiting, diarrhea, hyperbilirubinemia) occurring before day +100 posttransplant, with aGVHD staged by organ system and graded according to the modified Glucksberg criteria.26 L-aGVHD was defined according to the 2005 NIH-CC and included aGVHD manifestations occurring on or after day +100.20 Isolated hyperbilirubinemia occurring after day +100, in the absence of other cGVHD manifestations or an alternative etiology, was classified as L-aGVHD. This included cases in which a liver biopsy confirmed GVHD, because liver histopathology alone cannot differentiate between aGVHD and cGVHD.20
cGVHD assessment and central adjudication of cGVHD status
Transplant physicians with experience in the assessment of cGVHD (site principal investigators [PIs]) completed an initial-onset cGVHD CRF following clinical assessment of the patient around the time of cGVHD diagnosis, detailing the clinical manifestations and laboratory investigations of cGVHD according to the 2005 NIH-CC (supplemental Data, Initial Onset of cGVHD Case Report Forms).20 Usually, this CRF was completed before systemic immune suppression was escalated. Reasonable time was allowed to complete further investigations (eg, biopsies, PFTs) to establish a cGVHD diagnosis. Biopsies were recorded but not mandated. Centers were required to justify their cGVHD diagnosis according to the 2005 NIH-CC. Once submitted, initial-onset cGVHD CRFs were reviewed in near real-time by the study PI (G.D.E.C.). Given the insidious nature of cGVHD, if a cGVHD diagnosis was equivocal and/or the center was unable to justify the cGVHD diagnosis according to the NIH-CC, inquiries were sent to the site PI for further clarification. If resolution was not obtained, review by a study adjudication committee composed of experts in cGVHD evaluation (C.L.K., D.A.J., E.R.N., K.R.S., T.S., V.A.L., and J.T.W.) occurred. Final adjudication of GVHD status occurred by vote. As experience grew, as well as after reassessment by the local site PI and study committee, a subset of patients was still believed to possibly or likely have cGVHD based upon clinical and laboratory features, despite not completely meeting NIH-CC. These patients were included in the cGVHD group as “non–NIH-CC” cGVHD cases. Unequivocal cGVHD cases meeting NIH-CC were not reviewed by the study committee. L-aGVHD was identified from clinical CRFs submitted by sites, as well as from patients initially submitted as cGVHD cases, but who were agreed upon as better classified as L-aGVHD, upon further review with the site PI and/or study adjudication committee. Cases in which aGVHD features were present concurrently with ≥1 diagnostic and/or distinctive cGVHD manifestation (with appropriate investigations) were classified as overlap syndrome and included as cGVHD cases. A follow-up CRF at 12 months (±1 month) post-HSCT was also reported for cGVHD cases, documenting new cGVHD manifestations, organ-specific severity scoring, and maximal overall cGVHD global severity scoring according to NIH-CC in the first year post-HSCT.
Pulmonary cGVHD reassessment as per 2014 NIH-CC
A major change in the 2014 NIH-CC occurred with respect to diagnosing pulmonary cGVHD, including a decreased emphasis on lung biopsy to document pathological bronchiolitis obliterans (BO) and an increased emphasis on PFT findings.21 In the presence of another distinctive cGVHD manifestation, criteria that incorporated obstructive PFTs and computed tomography (CT) scans with air trapping, bronchiectasis, and/or small airway thickening could diagnose BO syndrome (BOS) and fulfill criteria for pulmonary cGVHD. To determine the utility of the 2014 NIH-CC criteria in children, centers that reported cases of pulmonary cGVHD were requested to provide additional information regarding the diagnosis (PFT results, CT scans, biopsy results).
Statistical analysis
See supplemental Data, Statistical Analysis.
Results
Patient characteristics
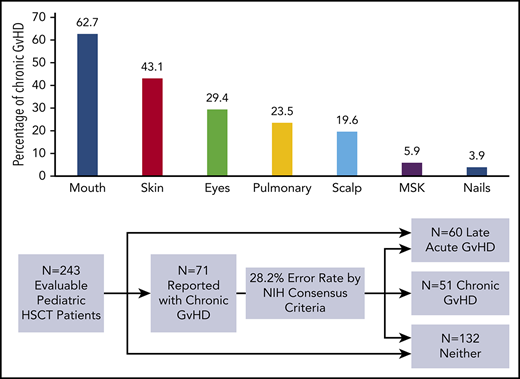
The 1-year event-free survival was 78.5% ± 2.4% and transplant-related mortality was 9.2% ± 1.7% for the entire cohort. Of the 302 enrolled patients, 243 (80.5%) were alive, engrafted, and disease-free at day +114 and evaluable for long-term GVHD status (Figure 1; Table 1). Two-hundred and twenty-seven (93.4%) completed follow-up to the end of study period, with the other 16 (6.6%) completing follow-up to a median day +244.5 (interquartile range, day 207-309.5) before experiencing relapse (n = 9) or late-toxic mortality (n = 7). Fifty-one (21% of the evaluable population) developed cGVHD at a median day +161 (interquartile range, day 129-203; range, day 41-385) (Figure 2), including 43 (84.3%) meeting NIH-CC for cGVHD and 8 (15.7%) being “non–NIH-CC” cGVHD cases. Sixty patients (24.7% of the evaluable population) had L-aGVHD only; the majority (80%) experienced persistence of or recurrence from aGVHD before day +100. Only 12 patients (4.9% of the evaluable population, or 20% of L-aGVHD cases) had true de novo L-aGVHD. The remaining 132 (54.3% of the evaluable population) had no evidence of L-aGVHD or cGVHD.
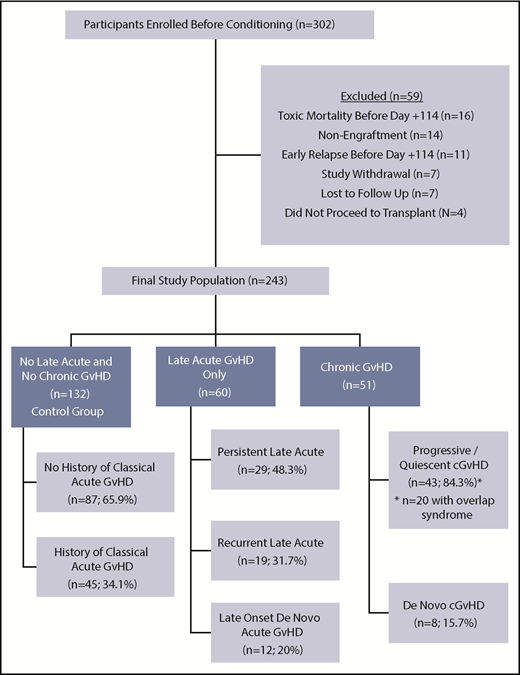
Flow diagram of enrolled patients into 3 final GVHD groupings according to NIH-CC. A proportion of patients in all 3 groups had a history of classical aGVHD before day +100, including 45 of 132 (34.1%) in the control group, 43 of 51 (84.3%) in the cGVHD group, and 48 of 60 (80%) in the L-aGVHD group. Of the 51 pediatric patients with cGVHD (43 meeting NIH-CC), most (84.3%) had progressive/quiescent cGVHD, meaning that their cGVHD developed concurrently with or after a past history of aGVHD. Twenty cGVHD cases (39.2%) had concurrent aGVHD manifestations at the time of cGVHD diagnosis (overlap syndrome), and 8 others (15.7%) had a history of L-aGVHD that had resolved before their quiescent cGVHD diagnosis. De novo cGVHD without a history of aGVHD was rare (15.7%). L-aGVHD cases were further subdivided into persistent L-aGVHD (aGVHD persisting after day +100; n = 29; 48.3%), recurrent L-aGVHD (aGVHD manifestations returning after day +100 following a period of resolution; n = 19; 31.7%), or new-onset de novo L-aGVHD (n = 12; 20%).
Flow diagram of enrolled patients into 3 final GVHD groupings according to NIH-CC. A proportion of patients in all 3 groups had a history of classical aGVHD before day +100, including 45 of 132 (34.1%) in the control group, 43 of 51 (84.3%) in the cGVHD group, and 48 of 60 (80%) in the L-aGVHD group. Of the 51 pediatric patients with cGVHD (43 meeting NIH-CC), most (84.3%) had progressive/quiescent cGVHD, meaning that their cGVHD developed concurrently with or after a past history of aGVHD. Twenty cGVHD cases (39.2%) had concurrent aGVHD manifestations at the time of cGVHD diagnosis (overlap syndrome), and 8 others (15.7%) had a history of L-aGVHD that had resolved before their quiescent cGVHD diagnosis. De novo cGVHD without a history of aGVHD was rare (15.7%). L-aGVHD cases were further subdivided into persistent L-aGVHD (aGVHD persisting after day +100; n = 29; 48.3%), recurrent L-aGVHD (aGVHD manifestations returning after day +100 following a period of resolution; n = 19; 31.7%), or new-onset de novo L-aGVHD (n = 12; 20%).
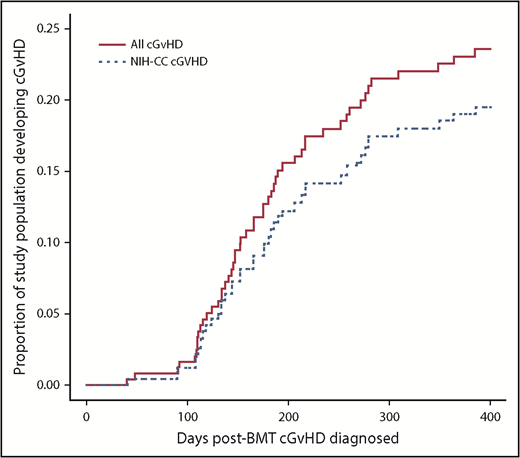
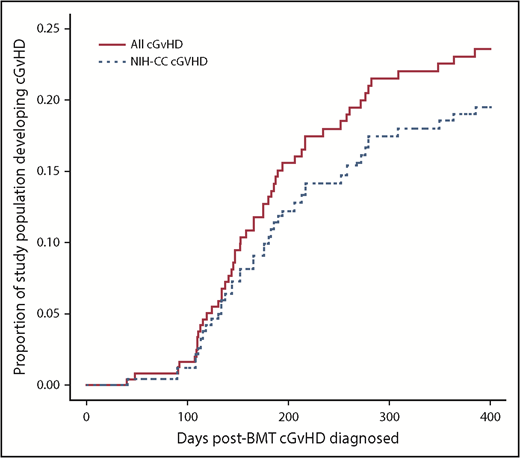
Time course for cGVHD diagnosis over the first-year posttransplant. Red solid line indicates all children diagnosed with cGVHD. Blue dotted line indicates only the children with NIH-CC cGVHD.
Time course for cGVHD diagnosis over the first-year posttransplant. Red solid line indicates all children diagnosed with cGVHD. Blue dotted line indicates only the children with NIH-CC cGVHD.
Central adjudication of GVHD clinical features improved cGVHD assessment
During the study period, inquiries were made by the study PI to the site PI (or vice versa) to clarify cGVHD clinical manifestations against the NIH-CC in 44 patients (supplemental Table 1). For 5 patients, communication led to resolution before official reporting as a cGVHD case (3 agreed most consistent with L-aGVHD, 2 deemed inconsistent with any form of GVHD). For the other 39 patients, inquiries occurred after sites submitted initial-onset cGVHD forms documenting the clinical manifestations of cGVHD to the study. For these, central review by the study committee occurred only when the reported GVHD manifestations remained equivocal for a cGVHD diagnosis following further review between the site PI and study PI, which occurred in 25 of 39 (64.1%) of submitted cases. Although 16 of 39 (41.0%) of the reported cGVHD cases were confirmed as meeting NIH-CC, 23 of 39 (59.0%) were agreed upon (by the study PI, site PI, and study committee) as not meeting NIH-CC, including 10 that were better classified as L-aGVHD, 5 that were felt not to be related to GVHD at all (because of their very mild severity, resolution without treatment, and/or alternative etiology later found), and 8 (all reviewed by the study committee) where it was believed that the participant probably or likely had cGVHD, despite the NIH-CC not being formally met (“non–NIH-CC” cGVHD cases). The most common reason for reclassification of submitted cGVHD cases involved patients with clinical manifestations of GVHD occurring after day +100 that were most consistent with L-aGVHD (eg, maculopapular rash, diarrhea, nausea and vomiting, and isolated hyperbilirubinemia) in the absence of other diagnostic or distinctive cGVHD manifestations. Stated differently, had the ABLE/PBMTC 1202 biomarker study relied only upon sites submitting dichotomous yes/no answers regarding the presence of cGVHD, the cGVHD group could have been as large as 71 patients, of whom 20 (13 L-aGVHD and 7 with signs and symptoms better ascribed to non-GVHD causes) would have been erroneously classified as having cGVHD (error rate, 28.2%).
Clinical manifestations of cGVHD in children at the time of diagnosis
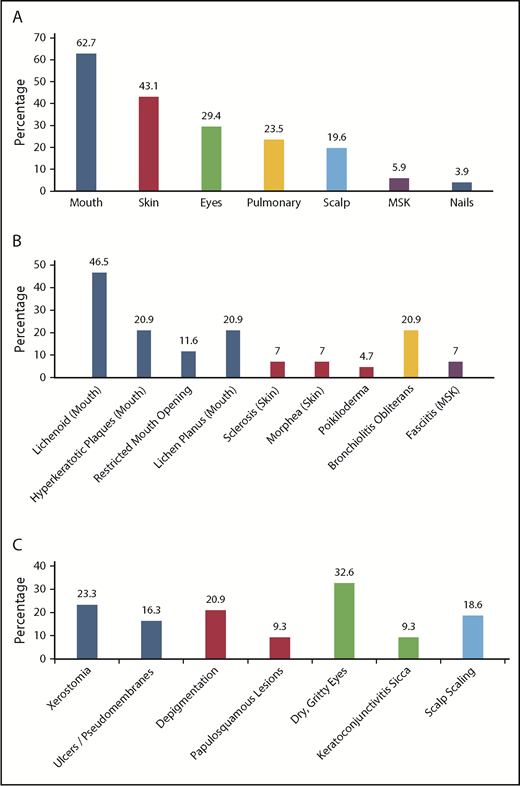
Of the 43 patients meeting NIH-CC for cGVHD, 36 (83.7%) exhibited ≥1 diagnostic cGVHD manifestation at the time of initial diagnosis. For the other 7, cGVHD diagnosis was established according to the NIH-CC by having ≥1 distinctive cGVHD sign along with supporting laboratory values or biopsy results (supplemental Table 2). An additional 8 patients (“non–NIH-CC cGVHD”) were believed to possibly (n = 6) or likely (n = 2) have cGVHD after study committee review, despite not meeting formal NIH-CC (supplemental Table 3). These “non–NIH-CC cGVHD” patients all had low NIH-CC organ severity scores (1-2), suggesting that mild presentation may have made the diagnosis according to the NIH-CC more challenging and/or the disease was diagnosed at an earlier time in the natural history of cGVHD development. The most frequent organ systems involved with diagnostic or distinctive cGVHD manifestations in children, in order of decreasing incidence, included the mouth, skin, eyes, lungs, scalp, musculoskeletal system, and nails (Figure 3). No patient was reported with diagnostic or distinctive cGVHD features of the gastrointestinal tract (esophageal webs or strictures) or genitalia (lichen planus features, vaginal scarring/stenosis, ulceration, erosions, or fissures) at diagnosis or follow-up. Because the study did not record whether gynecologic examinations were routinely performed, it is uncertain whether the absence of cGVHD of the genitalia is a true pediatric phenomenon or is due to the lack of a gynecologic evaluation.
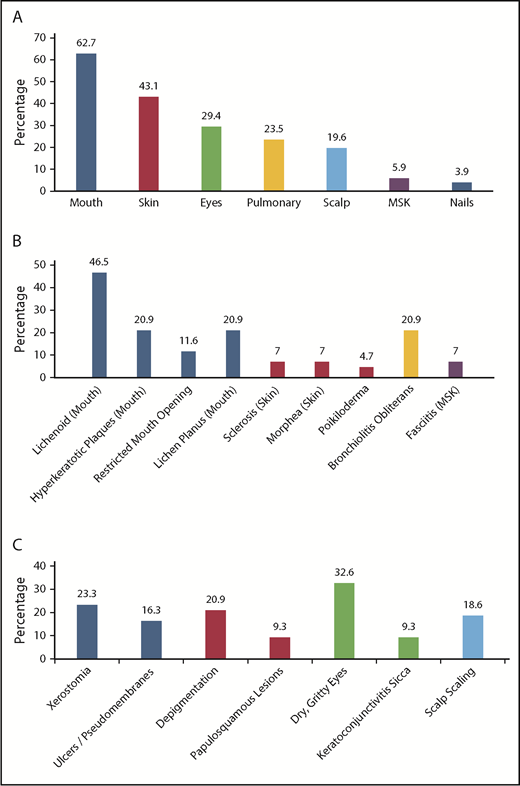
Organ systems and clinical manifestations of cGVHD in children at the time of initial cGVHD diagnosis. (A) The percentage of children with cGVHD (NIH-CC and non–NIH-CC) with specific organ system involvement. (B) The percentage of children with NIH-CC cGVHD exhibiting diagnostic cGVHD signs at the onset of cGVHD development. (C) The percentage of children with NIH-CC cGVHD exhibiting distinctive cGVHD signs at the onset of cGVHD development. MSK, musculoskeletal.
Organ systems and clinical manifestations of cGVHD in children at the time of initial cGVHD diagnosis. (A) The percentage of children with cGVHD (NIH-CC and non–NIH-CC) with specific organ system involvement. (B) The percentage of children with NIH-CC cGVHD exhibiting diagnostic cGVHD signs at the onset of cGVHD development. (C) The percentage of children with NIH-CC cGVHD exhibiting distinctive cGVHD signs at the onset of cGVHD development. MSK, musculoskeletal.
By comparison, common GVHD signs and symptoms (seen in aGVHD and cGVHD) and “other” features (rare, controversial, or nonspecific) frequently co-occurred in cGVHD cases, with 47 of 51 (92.2%) cGVHD patients exhibiting involvement in ≥1 organ system (Table 2). Such nonspecific signs and symptoms were present at the time of cGVHD diagnosis in 1 organ system in 13 patients (25.5%), 2 organ systems in 16 patients (31.4%), 3 organ systems in 9 patients (17.6%), 4 organ systems in 7 patients (13.7%), and 5 organ systems in 2 patients (4%). When nausea, vomiting, diarrhea, maculopapular skin rash, and/or hyperbilirubinemia occurred concurrently with other cGVHD manifestations, these patients were labeled as having overlap syndrome, which occurred in 20 of 51 (39.2%) patients in the cGVHD group.
Biopsies of involved tissues were reported for 16 cGVHD patients (skin [n = 8], gastrointestinal tract [n = 3], liver [n = 2], lungs [n = 2], renal [n = 1]). Six (37.5%) were inconclusive for a GVHD diagnosis. Three patients experienced early cGVHD manifestations before day +100 (onset at days +41, +90, and +92).
Global severity scoring of cGVHD in children
Sites scored each organ system involved with cGVHD for severity as per the NIH-CC, at initial diagnosis and at 12 months (±1 month) post-HSCT (or at time of death, if occurred first). New organ system involvement, with cGVHD developing after initial diagnosis, was recorded. Sites also provided their assessment of the maximal severity of cGVHD over the first year posttransplant, allowing for the calculation of the maximal NIH-CC cGVHD global severity score for each patient, a useful measure associated with nonrelapse mortality and survival,27,28 and the total impact of cGVHD on organ and functional status.20,21,29-31 Children with confirmed NIH-CC cGVHD were assessed at maximal severity as having mild cGVHD (6/43; 14%), moderate cGVHD (16/43; 37.2%), or severe cGVHD (21/43; 48.8%); the median number of organs involved was 3 (range, 1-5).
The 2014 NIH-CC inadequately address pulmonary cGVHD in children
We noticed particular challenges with diagnosing pulmonary cGVHD according to NIH-CC in children, despite pulmonary dysfunction being reported to the study. Additional information was sought from centers in an attempt to reclassify the 12 reported patients with pulmonary cGVHD against the 2014 NIH-CC, which place increased emphasis on PFTs as opposed to lung biopsy.21 Only 4 of 12 (33.3%) met formal 2014 NIH-CC for pulmonary cGVHD (pathological BO or BOS), with 2 meeting the criteria only after death from severe pulmonary dysfunction and autopsy confirmation of BO. Interestingly, both patients had undergone open lung biopsy while alive that were nondiagnostic. Only 2 of 12 (16.7%) met the 2014 NIH-CC for BOS while alive, both of whom were older (14 and 15.4 years) and able to reliably complete PFTs. Reasons for not meeting the 2014 NIH-CC were many, with >1 reason often present in the same individual (Table 3). Although it might be argued that these patients did not truly have pulmonary cGVHD, 9 of 12 met NIH-CC for a cGVHD diagnosis because of diagnostic and/or distinctive cGVHD manifestations in other organ systems, arguing that the respiratory syndrome was likely a component of pulmonary cGVHD. Only 2 patients had symptomatic pulmonary NIH organ severity scores of 1 (mild, shortness of breath with climbing 1 flight of steps), whereas 7 patients had severity scores of 2 (moderate, shortness of breath after walking flat ground), and 4 patients had severity scores of 3 (severe, shortness of breath at rest and/or requiring oxygen). At 12 months (±1 month) after HSCT, 3 had NIH-CC lung scores of 0 (suggesting resolution), 3 were stable (identical lung severity score relative to diagnosis), 1 had progressive pulmonary severity but was alive, 4 had died from progressive pulmonary failure, and 1 had died from relapsed malignancy.
Risk factors for cGVHD and L-aGVHD development in children
Univariable analysis revealed a significant increase in the risk of cGVHD with malignant disease (odds ratio [OR], 2.84; P = .0069), peripheral blood stem cell (PBSC) grafts (OR, 3.56; P = .007), unrelated donors (OR, 3.57; P = .0065), HLA mismatch (OR, 3.21; P = .003), recipient age ≥12 years at the time of transplant (OR, 2.2; P = .03), and a history of classical aGVHD (OR, 10.39; P < .001). Such risk was significantly reduced when using GVHD prophylaxis agents other than a calcineurin inhibitor with methotrexate (OR, 0.33; P = .0087). By comparison, only malignant disease (OR, 3.08; P = .0022), HLA mismatch (OR, 2.36; P = .031), and a history of classical aGVHD (OR: 7.73; P < .001) were risk factors for L-aGVHD by univariable analysis (supplemental Table 4). Multivariable analysis revealed that risk for cGVHD development in children increased with a history of classical aGVHD grades 2 to 4 (OR, 5.48; P = .0001), PBSC grafts (OR, 4.11; P = .0064), and recipient age ≥12 years (OR, 3.07; P = .015), whereas such risk was reduced when a GVHD prophylaxis regimen other than a calcineurin inhibitor with methotrexate was used (OR, 0.24; P = .0132). By comparison, only a history of classical aGVHD grades 2 to 4 (OR, 6.35; P < .0001) and PBSC grafts (OR, 3.13; P = .0271) were significantly associated with an increased risk of L-aGVHD on multivariable analysis (Table 4; supplemental Table 5).
Discussion
cGVHD is a heterogeneous disease in terms of clinical signs and symptoms, organ involvement, and severity. Accurate clinical assessment of cGVHD provides the foundation for all subsequent cGVHD research, whether related to biomarkers, registry studies, or therapeutic clinical trials.16,32 However, a number of studies clearly demonstrate inconsistency between clinicians with respect to cGVHD assessment, with wide interobserver variability in cGVHD diagnosis and severity scoring.33-36 A recent Blood and Marrow Transplant Clinical Trials Network multicenter clinical trial, for instance, involving the treatment of established cGVHD and occurring at experienced transplant institutions, excluded 10% of participants post hoc for not satisfying the NIH-CC for cGVHD diagnosis at the time of enrollment.37 Further, although registry studies regarding cGVHD provide access to large numbers of patients for statistical analysis, the retrospective data collection without adjudication of cGVHD diagnosis raises serious concern about the validity of the diagnosis to begin with. To address this, the Center for International Blood and Marrow Transplant Research implemented cGVHD reporting according to the 2014 NIH-CC on their research-level CRFs in 2016.38 However, multiple barriers to using the NIH-CC have been reported, including lack of time, unfamiliarity with the recommendations, controversy with the criteria, and requirement for investigator training, raising concerns about whether such data from a registry will be sufficient for analysis.34,38-40 These issues are likely compounded in children, in whom cGVHD is not encountered as frequently compared with adult allo-HSCT.
The ABLE/PBMTC 1202 study represents the largest study documenting the clinical features of cGVHD and L-aGVHD in children according to the NIH-CC. This study has a number of strengths, including the prospective data collection, requirement for sites to justify their cGVHD diagnosis according to the NIH-CC, the near real-time review of cGVHD cases, central review and adjudication of the cGVHD diagnosis, and the multi-institutional nature of the study, which included smaller to larger pediatric bone marrow transplantation centers, thus reflecting a real-world analysis. An important finding was that, in our attempt to meticulously document cGVHD in children according to the NIH-CC, 28.2% of submitted cGVHD cases were reclassified as something other than cGVHD. If extrapolated to larger registry data, this could imply that >1 in 4 documented cases of pediatric cGVHD may not truly be cGVHD. Therefore, we suggest that future studies of cGVHD in children, whether regarding biomarkers or clinical trials using cGVHD as an end point, require a judicious and detailed process for assessing and adjudicating cGVHD diagnoses. In our analysis, L-aGVHD in children was of similar incidence to cGVHD (24.7% vs 21%) and was the major reason to reclassify a diagnosis in submitted cGVHD cases. Indeed, manifestations common to aGVHD and cGVHD (as well as “other” features) were present in 92.2% of cGVHD cases, with true overlap syndrome (simultaneous aGVHD and cGVHD) occurring in 39.4% of cGVHD cases. In our experience, separating clinical manifestations as being acute or chronic was challenging for clinicians. However, as the trial continued, fewer reports of cGVHD needed to be reclassified as L-aGVHD, suggesting a positive learning curve in the context of a multi-institution study. Although in-depth training around assessing patients according to the NIH-CC was not provided at the start of the ABLE/PBMTC 1202 study, this will be an emphasis in our next cGVHD biomarker study in children (ABLE 2.0/PBMTC 1901), which will open in 2019. Our data also strengthen the link between acute and chronic GVHD, with very few children having true de novo cGVHD and the major risk factor for cGVHD being a history of grades 2 to 4 aGVHD.
Our study supports use of the NIH-CC for cGVHD evaluation in children. Although we used the 2005 NIH-CC as opposed to the 2014 NIH-CC (because the latter had not been published when the study opened), this should have minimal impact given that the diagnostic criteria changed little between the 2 versions.38 Importantly, 15.7% of our cGVHD cases did not meet NIH-CC (“non–NIH-CC cGVHD cases”); however, site PIs and the study committee agreed that the patient likely or probably had cGVHD. This is not surprising given that the NIH-CC were designed to identify patients with unequivocal cGVHD for enrollment in clinical trials, and it is accepted that many patients with clinical features of cGVHD and active alloreactivity (but not meeting NIH-CC) are encountered in clinical practice.38 However, we believe that there is still room for improvement in the cGVHD diagnostic criteria in future iterations of the NIH-CC, specifically for children. This is particularly true for pulmonary cGVHD; the pathologic diagnosis of BO by lung biopsy and the PFT criteria for BOS perform poorly, particularly in young children unable to adequately complete PFTs. New modalities, such as parametric response mapping from high-resolution inspiratory/expiratory CT scans, which quantify the degree of functional small airway disease relative to normal lung parenchyma, have shown promise in differentiating BOS from pulmonary infection without BOS and could offer a noninvasive means of diagnosing early pulmonary cGVHD in children without the need for PFTs.41 Cellular and plasma biomarkers, such as higher percentages of CD19+ CD21Low B cells and elevated B-cell activating factor,42 matrix metalloproteinase-3,43 and Krebs Von Den Lungen-6,44 have also been found to be potential biomarkers for pulmonary BOS following allo-HSCT, which could further aid in the early asymptomatic diagnosis of the disorder and act synergistically with other noninvasive diagnostic tests. Further, although the NIH-CC require the absence of infection to diagnose BOS, given the frequency of concurrent respiratory infections in children, this may not be possible. We believe that the NIH-CC for pulmonary cGVHD need to be simplified, and investigations required should be more in line with the developmental stage of young children.
When NIH-CC for cGVHD are met in children, the most common organ systems involved in our study at the time of diagnosis were the mouth, skin, eyes, and lungs. cGVHD of the musculoskeletal system (fasciitis) and sclerosis of the skin were relatively rare at first presentation, possibly reflecting that these manifestations represent a more terminal event in the natural history of cGVHD, once scarring and fibrosis set in. The most significant risk factors for cGVHD and L-aGVHD in children from our data were a history of aGVHD and PBSC grafts. Interestingly, recipient age ≥12 years was a risk factor for cGVHD, but not L-aGVHD, which might suggest that hormonal changes associated with puberty could influence immune dysregulation and alloreactivity associated with cGVHD. This hypothesis requires further study in future research regarding cGVHD in adolescents.
Given the longer life expectancy of children and the significant morbidity and cost associated with cGVHD, efforts to reduce cGVHD development in this population are imperative. Modifiable actions supported by our data might include the use of bone marrow over PBSCs as the graft source and judicious effort to prevent higher-grade 2 to 4 aGVHD. Other strategies increasingly used in pediatric transplant, including αβ T-cell receptor–depleted haploidentical transplant45,46 and posttransplant cyclophosphamide for GVHD prevention,47-49 have shown promise in children with malignant and nonmalignant disease, including low (and sometimes nonexistent) rates of cGVHD with excellent long-term survival outcomes. When cGVHD does occur, the NIH-CC can be used for diagnosing cGVHD in children; however, future iterations of the criteria should be modified to reflect pediatric-specific issues.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Ross Tsuyuki and Debbie Boyko (EPICORE, University of Alberta) for help with the operationalization of the ABLE/PBMTC 1202 study and Imran Hassan (EPICORE, University of Alberta) for statistical support with the initial biomarker studies. The authors also acknowledge all of the clinical research associates at each center who helped with data acquisition, as well as the Canadian Glycomics Network and, in particular, Stephanie Maier, for help with acquiring and interpreting ABO blood group data.
This work was supported by the Canadian Institutes of Health Research (grant 255075). Pediatric Blood and Marrow Transplant Consortium efforts in this study were supported in part by a National Institutes of Health, National Heart, Lung, and Blood Institute grant U01HL069254 and by a grant from the Johnny Christopher Fund/St. Baldrick's Foundation.
Authorship
Contribution: G.D.E.C designed the research and was a PI for the ABLE/PBMTC 1202 study, contributed patients to the study, was a member of the study committee, performed data analysis, and takes primary responsibility for writing of the manuscript; K.R.S. designed the research and was a PI for the ABLE/PBMTC 1202 study, was a member of the study committee, and helped to write and edit the manuscript; E.R.N., J.T.W., C.L.K., V.A.L., T.S., and D.A.J. were site PIs, contributed patients to the study, were members of the study committee, including cGVHD case adjudication, and provided writing and editorial review of the manuscript; A.C.H., M.A.P., H.B., S.W.C., E.H.C., K.A.K., M.B., B.R.O., A.F., S.C., D.C., J.H.C., M.J., S.S., A.B.P., G.C.M., D.M., A.C.C., and A.L. were site PIs, contributed patients to the study, and provided writing and editorial review of the manuscript; L.J.W. provided expertise regarding ABO blood group data; B.P. and Y.N.A.H. performed the statistical analyses and provided writing and editorial review of the manuscript; and A.H. was the primary study coordinator, aided with data collection between sites and the study, and provided writing and editorial assistance for the manuscript.
Conflict-of-interest disclosure: J.T.W. is employed by Pharmacyclics and was employed there for part of the study enrollment period. The remaining authors declare no competing financial interests.
Correspondence: Geoffrey D. E. Cuvelier, Manitoba Blood and Marrow Transplant Program, Pediatric Hematology-Oncology-BMT, CancerCare Manitoba, ON2011-675 McDermot Ave, Winnipeg, MB R3E 0V9, Canada; e-mail: gcuvelier@cancercare.mb.ca.