

Introduction: Although recent studies have refined the classification of B-progenitor and T-lineage acute lymphoblastic leukemia into gene-expression based subgroups, a comprehensive integration of significantly mutated genes and pathways for each subgroup is needed to understand disease etiology.
Methods: We studied 2789 children, adolescents and young adults (AYA) with newly diagnosed B-ALL (n=2,322 cases) or T-ALL (n=467) treated on Children's Oncology Group (n=1,872) and St. Jude Children's Research Hospital trials (n=917). The cohort comprised childhood NCI standard-risk (41.8%; age range 1-9.99 yrs, WBC ≤ 50,000/ml), childhood NCI high-risk (44.5%; age range ≥10 to 15.99 yrs) and AYA (9.9%; age range 16-30.7 yrs). Genomic analysis was performed on tumor and matched-remission samples using whole transcriptome sequencing (RNA-seq; tumor only; n=1,922), whole exome sequencing (n=1,659), whole genome sequencing (n=757), and single nucleotide polymorphism array (n=1,909).
Results: For B-ALL, 2104 cases (90.6%) were classified into 26 subgroups based on RNA-seq gene expression data and aneuploidy or other gross chromosomal abnormalities (iAMP21, Down syndrome, dicentric), deregulation of known transcription factors by rearrangement or mutation (PAX5 P80R, IKZF1 N159Y), or activation of kinase alterations (Ph+, Ph-like). For T-ALL, cases were classified into 9 previously described subtypes based on dysregulation of transcription factor genes and gene expression.
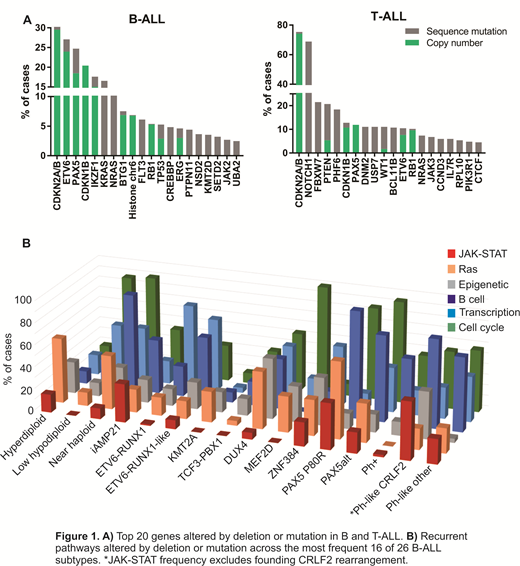
In 1,659 cases subject to exome sequencing (1259 B-ALL, 405 T-ALL) we identified 18,954 nonsynonymous single nucleotide variants (SNV) and 2,329 insertion-deletion mutations (indels) in 8,985 genes. Overall, 161 potential driver genes were identified by the mutation-significance detection tool MutSigCV or by presence of pathogenic variants in known cancer genes. Integration of sequence mutations and DNA copy number alteration data in B-ALL identified 7 recurrently mutated pathways: transcriptional regulation (40.6%), cell cycle and tumor suppression (38.0%), B-cell development (34.5%), epigenetic regulation (24.7%), Ras signaling (33.0%), JAK-STAT signaling (12.0%) and protein modification (ubiquitination or SUMOylation, 5.0%). The top 10 genes altered by deletion or mutation in B-ALL were CDKN2A/B (30.1%), ETV6 (27.0%), PAX5 (24.6%), CDKN1B (20.3%), IKZF1 (17.6%), KRAS (16.5%), NRAS (14.6%), BTG1 (7.5%) histone genes on chromosome 6 (6.9%) and FLT3 (6.1%), and for T-ALL, CDKN2A/B (74.7%), NOTCH1 (68.2%), FBXW7 (21.3%), PTEN (20.5%) and PHF6 (18.2%) (Figure 1A). We identified 17 putative novel driver genes involved in ubiquitination (UBE2D3, UBE2A, UHRF1, and USP1), SUMOylation (SAE1, UBE2I), transcriptional regulation (ZMYM2, HMGB1), immune function (B2M), migration (CXCR4), epigenetic regulation (DOT1L) and mitochondrial function (LETM1). We also observed variation in the frequency of genes and pathways altered across B-ALL subtypes (Figure 1B). Interestingly, alteration of SAE1 and UBA2, novel genes that form a heterodimeric complex important for SUMOylation, and UHRF1 were enriched in ETV6-RUNX1 cases. Deletions of LETM1, ZMYM2 and CHD4 were associated with near haploid and low hypodiploid cases. Deletion of histone genes on chromosome 6 and alterations of HDAC7 were enriched in Ph+ and Ph-like ALL. Mutations in the RNA-binding protein ZFP36L2 were observed in PAX5alt, DUX4 and MEF2D subgroups.
Genomic subtypes were prognostic. ETV6-RUNX1, hyperdiploid, DUX4 and ZNF384 ALL were associated with good outcome (5-yr EFS 91.1%, 87.2%, 91.9% and 85.7%, respectively), ETV6-RUNX1-like, iAMP21, low hyperdiploid, PAX5 P80R and PAX5alt were associated with intermediate outcome (5-yr EFS 68.6%, 72.2%, 70.8%, 77.0% and 70.9%, respectively), whilst KMT2A, MEF2D, Ph-like CRLF2 and Ph-like other conferred a poor prognosis (55.5%, 67.1%, 51.5% and 62.1%, respectively). TCF3-HLF and near haploid had the worst outcome with 5-yr EFS rates of 27.3% and 47.2%, respectively.
Conclusions: These findings provide a comprehensive landscape of genomic alterations in childhood ALL. The associations of mutations with ALL subtypes highlights the need for specific patterns of cooperating mutations in the development of leukemia, which may help identify vulnerabilities for therapy intervention.
Gastier-Foster:Bristol Myers Squibb (BMS): Other: Commercial Research; Incyte Corporation: Other: Commercial Research. Willman:to come: Patents & Royalties; to come: Membership on an entity's Board of Directors or advisory committees; to come: Research Funding. Raetz:Pfizer: Research Funding. Borowitz:Beckman Coulter: Honoraria. Zweidler-McKay:ImmunoGen: Employment. Angiolillo:Servier Pharmaceuticals: Consultancy. Relling:Servier Pharmaceuticals: Research Funding. Hunger:Jazz: Honoraria; Amgen: Consultancy, Equity Ownership; Bristol Myers Squibb: Consultancy; Novartis: Consultancy. Loh:Medisix Therapeutics, Inc.: Membership on an entity's Board of Directors or advisory committees. Mullighan:Amgen: Honoraria, Other: speaker, sponsored travel; Loxo Oncology: Research Funding; AbbVie: Research Funding; Pfizer: Honoraria, Other: speaker, sponsored travel, Research Funding; Illumina: Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: sponsored travel.

