Key Points
Avadomide monotherapy is well tolerated and demonstrates preliminary clinical efficacy in the treatment of R/R DLBCL patients.
Avadomide monotherapy resulted in mPFS of 6 months in classifier-positive vs 1.5 months in classifier-negative R/R DLBCL patients.

Abstract
Treatment options for relapsed/refractory (R/R) diffuse large B-cell lymphoma (DLBCL) are limited, with no standard of care; prognosis is poor, with 4- to 6-month median survival. Avadomide (CC-122) is a cereblon-modulating agent with immunomodulatory and direct antitumor activities. This phase 1 dose-expansion study assessed safety and clinical activity of avadomide monotherapy in patients with de novo R/R DLBCL and transformed lymphoma. Additionally, a novel gene expression classifier, which identifies tumors with a high immune cell infiltration, was shown to enrich for response to avadomide in R/R DLBCL. Ninety-seven patients with R/R DLBCL, including 12 patients with transformed lymphoma, received 3 to 5 mg avadomide administered on continuous or intermittent schedules until unacceptable toxicity, disease progression, or withdrawal. Eighty-two patients (85%) experienced ≥1 grade 3/4 treatment-emergent adverse events (AEs), most commonly neutropenia (51%), infections (24%), anemia (12%), and febrile neutropenia (10%). Discontinuations because of AEs occurred in 10% of patients. Introduction of an intermittent 5/7-day schedule improved tolerability and reduced frequency and severity of neutropenia, febrile neutropenia, and infections. Among 84 patients with de novo R/R DLBCL, overall response rate (ORR) was 29%, including 11% complete response (CR). Responses were cell-of-origin independent. Classifier-positive DLBCL patients (de novo) had an ORR of 44%, median progression-free survival (mPFS) of 6 months, and 16% CR vs an ORR of 19%, mPFS of 1.5 months, and 5% CR in classifier-negative patients (P = .0096). Avadomide is being evaluated in combination with other antilymphoma agents. This trial was registered at www.clinicaltrials.gov as #NCT01421524.
Introduction
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of non-Hodgkin lymphoma (NHL) globally,1 with estimated annual incidences of 3 to 7/100 000 in the United States and Europe.2,3 The majority of newly diagnosed DLBCL patients achieve a complete response (CR) after a standard regimen of rituximab in combination with chemotherapy, usually cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).4 However, 40% of DLBCL patients relapse or have primary refractory disease. Only 50% of patients who relapse are eligible for high-dose chemotherapy and autologous stem cell transplantation (ASCT); the majority of these do not proceed to transplant or are not cured after ASCT.5-9 Patients with refractory DLBCL, defined as progressive or stable disease as best response to chemotherapy or relapse ≤12 months post-ASCT, have a poor prognosis, with median overall survival (OS) of 4 to 6 months.10-13 In patients with relapsed/refractory (R/R) disease who are not candidates for potentially curative therapies, including anti-CD19 chimeric antigen receptor T-cell therapy,14 DLBCL remains a high unmet need for which new treatment options are necessary.
DLBCL is clinically and biologically heterogeneous.15-17 Gene expression profiling has identified 2 molecular subtypes corresponding to cell-of-origin (COO) markers: germinal center B-cell–like (GCB) and activated B-cell–like (ABC) DLBCL.18-20 The COO classification is prognostic in frontline DLBCL, with 5-year OS rates ranging from 30% to 56% and 60% to 78% for ABC- vs GCB-DLBCL, respectively.18,21,22 Although the prognostic significance of COO in R/R DLBCL is unclear,23-25 its role in predicting response to different classes of novel agents, such as lenalidomide26 or ibrutinib,27 may be important in patient selection for a given therapy. In addition to COO, characteristics of the tumor microenvironment21,28-31 and baseline counts of circulating immune cells32,33 have been associated with outcome. These findings are supported by studies indicating that DLBCL is a disease of immune dysfunction in which the tumor microenvironment contributes to growth of malignant cells and evasion of immune surveillance.34-36
Avadomide (CC-122) is a cereblon-modulating agent that exerts direct cell-autonomous activity against malignant B cells and immunomodulatory effects.37 Upon binding to cereblon, a substrate receptor in the cullin4 E3 ligase complex, avadomide promotes recruitment, ubiquitination, and subsequent proteasomal degradation of the hematopoietic transcription factors Ikaros (IKZF1) and Aiolos (IKZF3).38-40 Degradation of Ikaros and Aiolos leads to decreased proliferation and increased apoptosis of malignant B cells and costimulatory effects in T and natural killer (NK) cells.41-43 In preclinical models, avadomide has demonstrated antitumor activities in both ABC- and GCB-DLBCL cell lines, indicating that its activity is independent of COO.37
CC-122-ST-001 (NCT01421524) is a multicenter, open-label phase 1 study to evaluate the safety and preliminary efficacy of avadomide in patients with advanced tumors unresponsive to standard therapies. Results of dose escalation in patients with solid tumors and hematologic malignancies demonstrated acceptable safety and tolerability of avadomide, defined the nontolerated dose and maximum tolerated dose (MTD) on a continuous dosing schedule, provided evidence supporting use of Aiolos as a pharmacodynamic biomarker, and showed signs of clinical activity in NHL.44,45 Based on preliminary efficacy results from the dose-escalation portion of the trial, expansion cohorts in several tumor types, including glioblastoma, hepatocellular carcinoma, multiple myeloma, and NHL were studied.
We report the safety and clinical efficacy of avadomide monotherapy, based on continuous and intermittent dosing schedules, in patients with R/R DLBCL. We also describe the pharmacodynamic effects of avadomide on immune and tumor cell biomarkers and the correlation of a novel tumor microenvironment gene expression classifier with clinical activity. A companion publication by Risueño et al46 describes development of the gene expression classifier as a basis for patient selection and the biology associated with the classifier.
Patients and methods
Patients
Eligible patients were ≥18 years of age and had histologically confirmed DLBCL with progression on or inability to tolerate ≥1 prior anthracycline- or alkylating agent-containing regimen (with or without anti-CD20). Patients had measurable disease by International Working Group 2007 Revised Response Criteria for NHL (≥1 objectively measurable lesion),47 Eastern Cooperative Oncology Group performance status (ECOG PS) ≤2, absolute neutrophil count ≥1.5 × 109/L, platelets ≥60 × 109/L, and adequate hepatic and renal function.
The study was conducted according to the Declaration of Helsinki and International Council on Harmonization. The research ethics boards of all participating institutions approved the protocol. All patients provided written informed consent.
Study design and treatment
The MTD of avadomide hydrochloride (HCl), active-ingredient-in-capsule, was previously established for continuous oral dosing at 3 mg daily in multitumor cohorts.45 The current study was designed to evaluate the effects of continuous vs intermittent dosing schedules on safety and tolerability and to determine the recommended phase 2 dose. Enrollment into a cohort was to be closed and defined a nontolerated dose if dose-limiting toxicities (DLTs) as defined below were observed in ≥33% of enrolled patients within the first cycle or if the safety review committee independently assessed a dose level/schedule was not tolerable. A diagrammatic figure representing enrollment of independent cohorts and decisions for each cohort enrolled is presented in supplemental Figure 1 (available on the Blood Web site). Twenty-four DLBCL patients and 1 patient with mantle cell lymphoma received avadomide HCl daily on a continuous schedule (3 mg, 28 of 28 days). Avadomide HCl was administered to 47 patients on intermittent schedules (4 mg, 5/7 days [n = 39]; 4 mg, 21/28 days [n = 3]; and 5 mg, 5/7 days [n = 5]). Another 26 patients received avadomide in free-base form as a formulated capsule, administered on an intermittent schedule (3 mg on 5/7 days [n = 18] and 4 mg on 5/7-d [n = 8]). Dose escalation on the intermittent schedule was evaluated at a starting dose of 4 mg of avadomide HCl and 3 mg of avadomide formulated capsule.45 Each treatment cycle was 28 days, with no rest period between cycles. Treatment was administered until disease progression, unacceptable toxicity, or patient/physician decision to discontinue treatment, including withdrawal to receive intensive consolidation (ASCT or allogeneic stem cell transplantation).
Study end points and evaluations
Adverse events (AEs) were assessed using NCI-CTCAE (version 4.0). Tumor assessments were performed at screening and between days 15 and 28 of even-numbered cycles through cycle 6 and every 3 cycles thereafter; clinical responses were determined by investigator assessment per 2007 Revised Response Criteria for NHL.47 Dose-limiting toxicities were defined as clinically relevant AEs related to avadomide that began ≤28 days within the first dose (cycle 1), based on criteria previously described.45 Patients were evaluable for DLT if, during cycle 1 dosing, they had received ≥1 dose of avadomide and had experienced a drug-related DLT. Patients who did not experience a DLT were only considered DLT-evaluable if they had received ≥75% of the planned avadomide doses at the cohort-specified dose/schedule and had sufficient data for safety evaluation.
See supplemental Materials and methods for additional details.
Results
Patients
From 7 May 2013 to 20 December 2016, 97 patients with R/R DLBCL were enrolled. As of the 5 April 2018 data cutoff, 2 patients remained on therapy. Table 1 provides baseline characteristics. The median age was 62 years (range, 25-91 years), 36% were ≥65 years old, and 57% were male. Among all patients enrolled, 84 had de novo DLBCL and 12 had transformed lymphoma. Most had an ECOG PS of 0 or 1; 6% had an ECOG PS of 2. Most (65%) had previously received ≥3 lines of systemic anticancer therapy (median, 3; range, 1-13). Eighty-nine patients had previously received R-CHOP or equivalent anti-CD20 containing intensive chemotherapy; 50 patients (49% overall) had primary refractory disease, defined as <CR after R-CHOP. Eighteen patients (19%) had received ≥1 prior ASCT, 14 of whom (14% overall) experienced relapse within 12 months. In total, 64% of patients had chemorefractory DLBCL, defined as stable or progressive disease as best response to last chemotherapy or relapse ≤12 months post-ASCT.12
Avadomide treatment alters peripheral T-cell phenotypes and increases trafficking of macrophages and T cells to the tumor
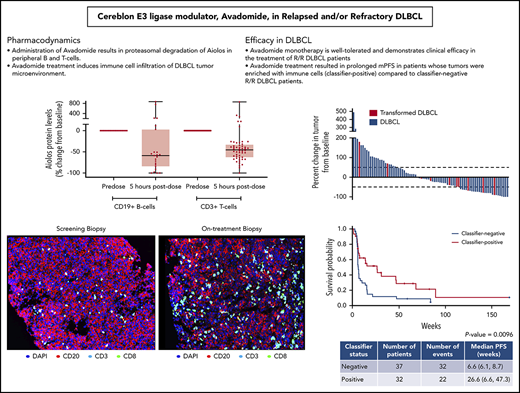
Cereblon-mediated degradation of Aiolos and Ikaros results in decreased malignant B-cell proliferation and activation of immune cells, such as T and NK cells.37,41,45,48 Analysis of peripheral blood samples collected 5 hours after administration of avadomide on C1D1 revealed that Aiolos levels in CD19+ B and CD3+ T cells were reduced by a median 59% and 45%, respectively, compared with predose levels (Figure 1A). Between C1D1 and C1D15, peripheral B-cell counts showed a precipitous decline (median, −81%) (supplemental Figure 2B).37,45 T cells in peripheral blood were activated at 1.5 hours postdosing on C1D1, as indicated by increased secretion of interferon γ (IFN-γ) (185%, P = .003) and interleukin-2 (300%, P = .018) in response to CD3-stimulation ex vivo (Figure 1B; supplemental Figure 2A), consistent with preclinical findings.41 Within 2 weeks of initiating avadomide, peripheral T-cell subsets showed a significant shift toward a more activated phenotype. Between baseline and C1D15, naive (CD45RA+/CD45RO−) CD8+ T cells declined by 30% (P = .0148), whereas memory (CD45RA−/CD45RO+) and activated (HLA-DR+) CD8+ T cells increased by 214% (P = .0006) and 111% (P = .001), respectively (Figure 1C). Similar shifts were observed in CD4+ T-cell subsets (supplemental Figure 2B). Absolute cell counts for total CD3+, CD4+, and CD8+ T-cell populations did not show marked changes from baseline (Figure 1C; supplemental Figure 2B).
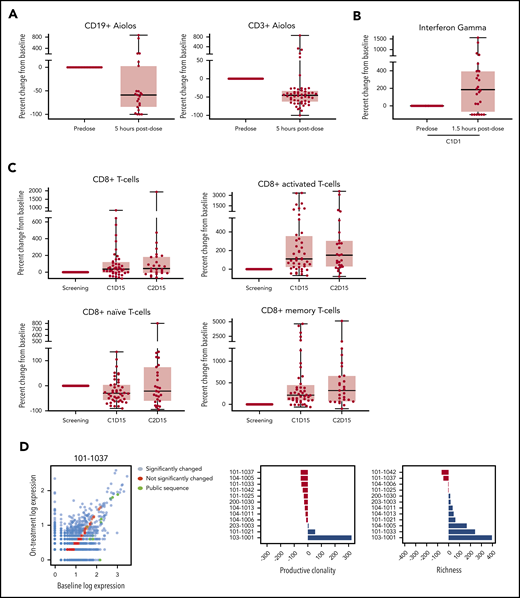
Avadomide effects on immune activation in peripheral blood. (A) Aiolos expression in peripheral CD19+ B and CD3+ T cells. (B) Ex vivo IFN-γ production in response to anti-CD3 stimulation. (C) Total peripheral blood CD8+ T cells and shifts in CD8+ T-cell subsets (naive, activated, memory). (D) TCR-β expression levels (log10-transformed number of CDR3 reads) at baseline vs on-treatment are plotted. Color indicates a significant differential expression (red, not significant; blue, significant; adjusted P < .001) or publicly identified TCR sequence with known antigen (green). Changes in TCR-β productive clonality and richness from baseline to end of cycle 2 are shown for each patient. Extent of change in percentage of baseline (0 indicating no change) is plotted.
Avadomide effects on immune activation in peripheral blood. (A) Aiolos expression in peripheral CD19+ B and CD3+ T cells. (B) Ex vivo IFN-γ production in response to anti-CD3 stimulation. (C) Total peripheral blood CD8+ T cells and shifts in CD8+ T-cell subsets (naive, activated, memory). (D) TCR-β expression levels (log10-transformed number of CDR3 reads) at baseline vs on-treatment are plotted. Color indicates a significant differential expression (red, not significant; blue, significant; adjusted P < .001) or publicly identified TCR sequence with known antigen (green). Changes in TCR-β productive clonality and richness from baseline to end of cycle 2 are shown for each patient. Extent of change in percentage of baseline (0 indicating no change) is plotted.
Given the observed increase in memory T cells and decrease in naive T cells and that increased T-cell diversity has been associated with clinical benefit in other malignancies, such as breast cancer,49 changes in T-cell clonality were explored through deep sequencing of the recombined V(D)J region of the T cell receptor β chain (TCR-β).50 Analysis of aligned sequences demonstrated both a significant expansion and contraction of specific clones in samples collected at the end of cycle 2 compared with baseline (Figure 1D). Analysis of aggregate data from 12 patients revealed an overall decrease in T-cell clonality and increased richness/diversity of identified T-cell clones. Given the sample size, it is difficult to make correlations to clinical outcome; however, the increased diversity may be a general pharmacodynamic effect of avadomide treatment and reflective of the ability of T cells to be reactivated. In addition, longitudinally collected viably frozen peripheral blood mononuclear cells were interrogated by mass cytometry to detect changes in other immune cell populations. Peripheral blood mononuclear cells collected on C1D22 had a median threefold increase (P = .001) in CD3+CD4+CD25+CD127−/loFoxP3+ regulatory T (Treg) cells compared with baseline (supplemental Figure 2C), although the functional implications of increased peripheral Treg cell counts are unknown. Additionally, a median decline of 95% in peripheral CD16+ monocytes (P ≤ .001) on C1D22 compared with baseline was observed (supplemental Figure 2D).
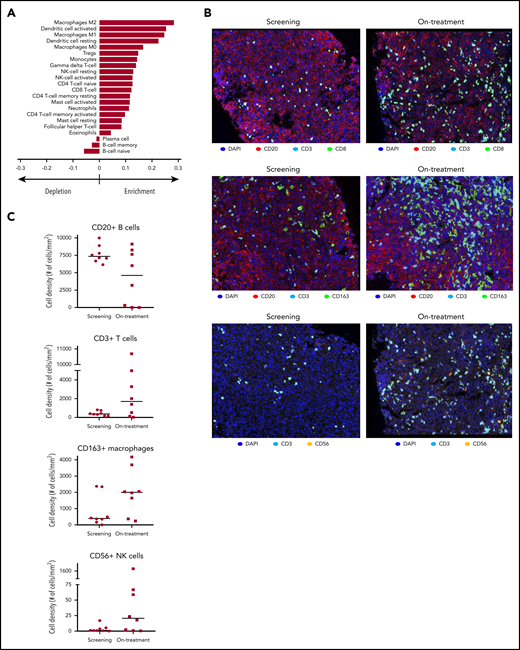
To understand the effects of avadomide on the tumor microenvironment, subsets of paired fresh-frozen tumor biopsy specimens obtained from patients (n = 18) at screening and 2 weeks posttreatment initiation were examined for changes in gene expression and immune cell composition. Analysis of bulk RNA-sequencing data using computational immune deconvolution algorithms revealed an enrichment of multiple T-cell populations, dendritic cells, and macrophages and a diminution in B-cell populations in on-treatment compared with screening biopsies (Figure 2A). Given the observed decline in peripheral monocytes, an increased abundance of macrophages in on-treatment biopsies may reflect greater trafficking of monocytes to the tumor with treatment. Gene set enrichment analysis51 revealed increased expression of genes associated with IFN-α responses (adjusted P = .04), indicative of an elevation in type I/II IFN production by cells such as T and NK cells in on-treatment vs screening biopsies (supplemental Figure 2E). Additionally, gene set enrichment analysis identified decreased expression in the E2F transcription factor pathway (adjusted P = .007), consistent with reduced proliferation of malignant B cells. This observation is supported by in vitro studies demonstrating avadomide-mediated inhibition of DLBCL B-cell proliferation.37
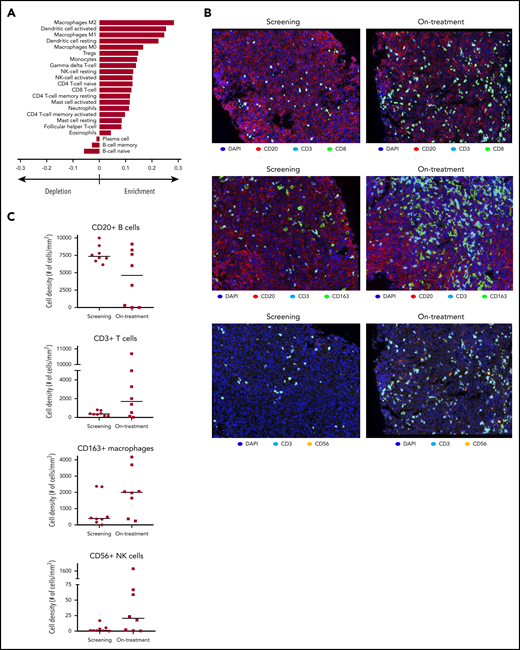
Avadomide effects signaling pathways and immune cell infiltration in DLBCL tumors. (A) RNA sequencing with immune cell deconvolution performed on screening and on-treatment (C1D10/15) biopsy specimens, showing enrichment/depletion relative to screening biopsy. (B) Cellular composition in paired tumor biopsy specimens by immunohistochemistry. Original magnification, ×20. (C) Intratumoral cell counts in screening vs on-treatment biopsy specimens by immunohistochemistry (CD20+ B cells; CD3+ T cells; CD163+ macrophages; CD56+ NK cells). DAPI, 4′,6-diamidino-2-phenylindole.
Avadomide effects signaling pathways and immune cell infiltration in DLBCL tumors. (A) RNA sequencing with immune cell deconvolution performed on screening and on-treatment (C1D10/15) biopsy specimens, showing enrichment/depletion relative to screening biopsy. (B) Cellular composition in paired tumor biopsy specimens by immunohistochemistry. Original magnification, ×20. (C) Intratumoral cell counts in screening vs on-treatment biopsy specimens by immunohistochemistry (CD20+ B cells; CD3+ T cells; CD163+ macrophages; CD56+ NK cells). DAPI, 4′,6-diamidino-2-phenylindole.
Results of the immune deconvolution analysis were confirmed by multiplex immunofluorescence enumeration of immune cell subsets in paired screening and on-treatment biopsies (n = 8). After administration of avadomide, there were trends toward increased numbers of infiltrating CD3+ T cells (median 360%), CD8+ T cells (924%), CD3+FOXP3+ Treg cells (321%), CD163+ macrophages (407%), CD163+CMAF+ M2 macrophages (579%), and CD11c+ dendritic cells (45%) and a decline in B cells (median, −38%) (Figure 2B-C; supplemental Figure 3A-B). Taken together, these results demonstrate that avadomide monotherapy exerts immunomodulatory effects, including increased numbers of T cells, NK cells, and macrophages and a decrease in proliferating B cells within the tumor.
Avadomide exposure, safety, and DLT
Following administration of a single oral dose of 3 mg in healthy adult subjects (n = 18), avadomide HCl and avadomide formulated capsule were absorbed with maximum plasma concentrations of 61.92 and 83.96 ng/mL and total plasma exposures (area under the curve ∞) of 666.34 and 774.86 h·μg/L, respectively (data not shown). The mean apparent terminal half-lives (t1/2) were similar (9.28 and 8.39 hours for avadomide HCl and formulated capsule, respectively). The mean apparent total plasma clearance was higher from avadomide HCl than the formulated capsule (4.50 vs 3.87 L/h), as was the mean apparent terminal volume of distribution (60.30 vs 46.86 L, respectively). However, after correction to the amount of avadomide free-base in the 2 formulations, the mean apparent total plasma clearance and mean apparent terminal volume of distribution were similar in the formulations.
Patient exposure to avadomide for each cohort is shown in supplemental Table 1. Median duration of avadomide treatment of the safety population in aggregate was 56 days (range, 7-1240 days). Avadomide MTD was established on a continuous schedule in the dose-finding part of the study (part A).45 During the initial part B dose expansion in DLBCL patients, neutropenia emerged as a common toxicity (Table 2). To mitigate neutropenia, intermittent schedules of 21/28 days and 5/7 days were tested. The 5/7-day schedule was introduced based on preclinical data from an in vitro assay of neutrophil differentiation.52 In these experiments, continuous treatment of bone marrow precursor cells with avadomide led to an arrest in myeloid maturation at a promyelocyte stage with a 90% decrease in mature neutrophil counts compared with vehicle control (supplemental Figure 4). Shifting to a 5/7-day treatment schedule partially relieved the arrest and allowed mature neutrophil populations in the cultures to expand. In patients, an intermittent (5/7-day) dosing schedule improved tolerability and permitted increased dose intensity, from 16.5 mg/week with 3 mg avadomide HCl continuously dosed vs 19 mg/week with 4 mg avadomide HCl on 5/7 days (supplemental Table 1). Importantly, the 5/7-day schedule permitted higher relative dose intensities (94% and 99% with 4 mg avadomide HCl and 3 mg formulated capsule, respectively) compared with continuous dosing (79% with 3 mg avadomide HCl). Thirty-five (36%) patients had ≥1 dose reduction, 89% of which was due to AEs (supplemental Table 2). Dose reductions occurred more frequently in patients receiving 3 mg avadomide HCl on a continuous schedule (44%) than in those receiving 4 mg avadomide HCl (36%) or 3 mg avadomide formulated capsule (11%) on 5/7 days. Of the 65 (67%) patients who experienced a dose interruption, 58 (60%) had the interruption in cycle 1 or 2. Incidence of dose interruption was not greatly affected by dosing schedule (64% for 3 mg avadomide HCl continuously dosed, 69% and 61% for 4 mg avadomide HCl and 3 mg formulated capsule, respectively, on 5/7 days). However, the median number of days of dose interruption due to AEs was higher in patients treated continuously with 3 mg avadomide HCl (14 days; range, 1-144) than in those treated on 5/7 days with 4 mg avadomide HCl (11 days, range 1-94) or 3 mg avadomide formulated capsule (9 days; range, 1-80). In addition, use of growth factors (granulocyte colony-stimulating factor, filgrastim, lenograstim, or pegfilgrastim) to mitigate severity of neutropenia was reduced in patients treated on a 5/7-day schedule (36% and 56% with 4 mg avadomide HCl and formulated capsule, respectively) compared with those dosed continuously (72% with 3 mg avadomide HCl). Fourteen of 83 DLT-evaluable patients (17%) experienced DLTs in cycle 1, leading to dose reduction and/or interruption in 10 individuals.
Among the most common (≥10%) TEAEs of any grade (Table 2; supplemental Table 2), the top 3 were neutropenia (66%), infections (57%), and asthenia (46%). Eighty-two patients (85%) had ≥1 grade 3/4 TEAEs; the most frequently reported were neutropenia (51%), infections (24%), anemia (12%), and febrile neutropenia (10%). Shifting from continuous dosing with 3 mg avadomide HCl to a 5/7-day schedule with 4 mg avadomide HCl or 3 mg avadomide formulated capsule was associated with a trend toward less frequent neutropenia (all grades: 76% vs 62% and 56%, respectively), less severe neutropenia (grades 3/4: 64% vs 39% and 39%, respectively), and less frequent febrile neutropenia (all grades: 12% vs 5% and 0%, respectively). During this study, 25 patients died. Twenty-one deaths resulted from disease progression; 3 deaths were due to other causes unrelated to study drug. One grade 5 AE of pneumonia was suspected to be related to avadomide, as reported by the investigator, who also attributed death of this patient to malignant lymphoma. The most common reason for treatment discontinuation (68%) was disease progression (supplemental Table 3). Five patients discontinued treatment when they became eligible for stem cell transplantation.
Efficacy
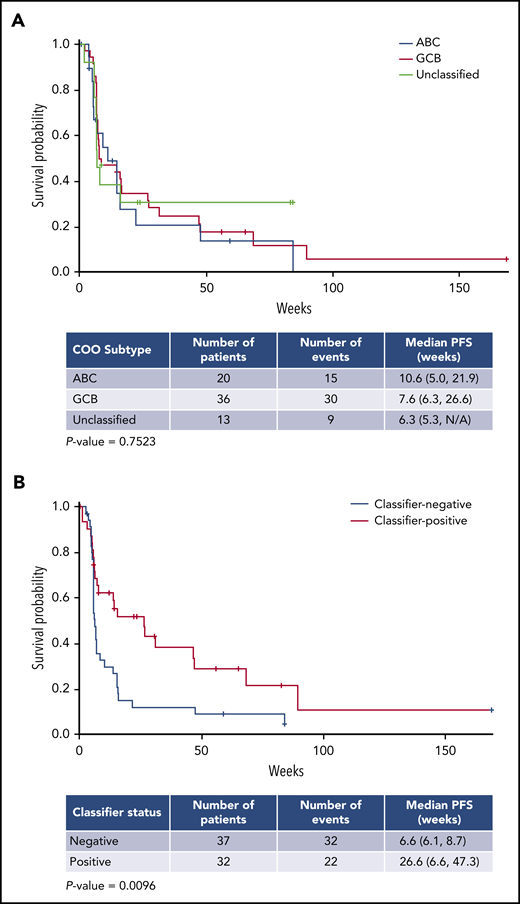
As of April 2018, the overall response rate (ORR) for all treated R/R DLBCL patients (n = 97) was 28%, with 9 patients achieving a CR (Table 3). Patients with de novo DLBCL had better outcomes than those with transformed lymphoma, as indicated by 6-month PFS rates of 28% vs 8% and CR of 11% vs 0%, respectively. Changes in lesion size from baseline were similar in de novo DLBCL vs transformed, possibly reflecting the low numbers of patients with transformed lymphoma (Figure 3). Clinical activity was observed in both the GCB- and ABC-DLBCL patient populations as defined by the NanoString lymphoma subtyping test (Table 3), supporting COO-independent activity, as predicted in preclinical studies.37 Among 84 DLBCL patients excluding transformed lymphoma, the ORR in GCB and ABC subtypes were 33% and 25%; the 6- and 12-month PFS rates in GCB vs ABC were 35% vs 21% and 18% vs 14%, respectively (Table 3). Moreover, COO was not correlated with the PFS rate in de novo DLBCL patients receiving avadomide therapy, as shown in a survival analysis (Figure 4A). Importantly, efficacy was not compromised with the intermittent 5/7-day schedule (data not shown).
![Best percentage reduction in target lesion burden over baseline in all response-evaluable patients. Dotted line indicates 50% reduction, consistent with a response (per investigator assessment, according to revised 2007 International Working Group [IWG] criteria). Blue, DLBCL de novo; red, transformed lymphoma.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/13/10.1182_blood.2019002395/7/m_bloodbld2019002395f3.png?Expires=1767761384&Signature=EBoj5I8wABbqsTkqkkhNzXh9t8hwsBBOwTIG3tysoMyz2TeEz8KF-Z-EVW9shYuoUyJwFjedE2mzdQ-QSB9iX6Z-ogCffbXSEQ3E2PVHludIfs6SrBWmyL4VKxhWpNdvtsdQyWhe8f--kqgKCab5Wo2hqnwk~CHaE14YnPMyoPSdv~-Kb8D5dv5YukgAKdCmpMq97d~dz6Rsr-aI~z~vTcK9XLyel9-PF69f6lyp9sUfOcyzYyUbl4vVPB6sIbThg~IaM8AU6eZJw1uCo59lCZfTv~ix7iKUxUI81radn6tsSsCPT3R0eBEswB2uuseDRdoKWZ-qEOvsSpWV~wkulw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Best percentage reduction in target lesion burden over baseline in all response-evaluable patients. Dotted line indicates 50% reduction, consistent with a response (per investigator assessment, according to revised 2007 International Working Group [IWG] criteria). Blue, DLBCL de novo; red, transformed lymphoma.
Best percentage reduction in target lesion burden over baseline in all response-evaluable patients. Dotted line indicates 50% reduction, consistent with a response (per investigator assessment, according to revised 2007 International Working Group [IWG] criteria). Blue, DLBCL de novo; red, transformed lymphoma.
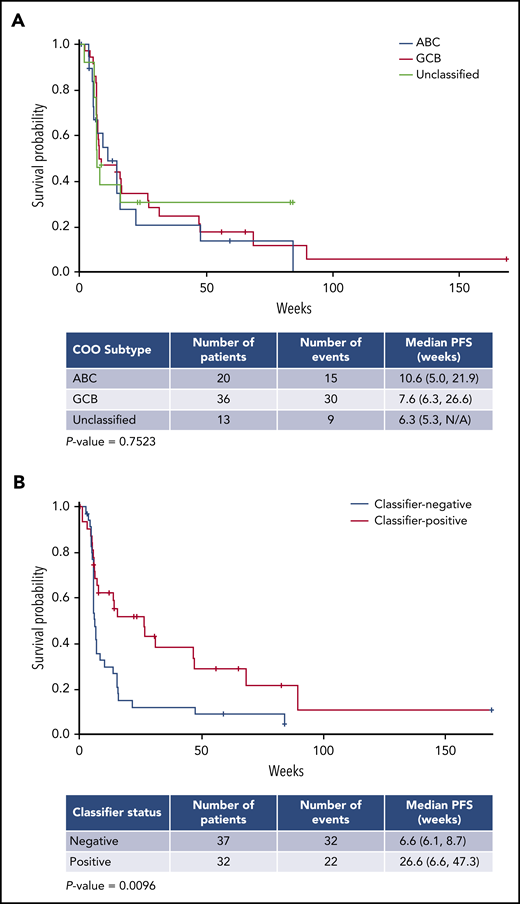
Progression-free survival (PFS) in de novo R/R DLBCL. PFS by COO (A) and gene expression classifier status (B). PFS was assessed by investigators. Asterisks represent censored observations. N/A, not available.
Progression-free survival (PFS) in de novo R/R DLBCL. PFS by COO (A) and gene expression classifier status (B). PFS was assessed by investigators. Asterisks represent censored observations. N/A, not available.
A novel gene expression classifier was developed via interrogation of a publicly available gene expression dataset21 to distinguish 2 patient subgroups in DLBCL,46 based on characteristics of the tumor microenvironment and in a manner independent of COO. One subgroup (classifier positive) is characterized by an enriched infiltration of T cells and macrophages, and the other subgroup (classifier negative) is associated with a predominance of intratumoral B cells. Avadomide’s dual mechanism of action, direct cell-autonomous anti-DLBCL and enhanced immunomodulatory activities, led to the hypothesis that patients with classifier-positive tumors would be more likely to respond clinically to avadomide than those with classifier-negative tumors. This hypothesis was tested by retrospectively assessing diagnostic biopsy specimens obtained prior to enrollment. Among 32 classifier-positive DLBCL patients not including transformed lymphoma, the ORR was 44%, mPFS was 27 weeks, and 6- and 12-month PFS rates were 52% and 29%, respectively, compared with classifier-negative patients, who had an ORR of 19%, mPFS of 7 weeks, and 6- and 12-month PFS rates of 12% and 9%, respectively (Table 3). A reduction in lesion size from baseline was associated with classifier status, although this association did not reach significance (data not shown). Among all avadomide-treated patients with de novo DLBCL, classifier-positive status was found to be significantly associated with PFS rate (mPFS Cox proportional hazard ratio, 0.490; 95% confidence interval, 0.280, 0.857; log-rank P = .0096), supporting its potential value as an enrichment strategy for avadomide response (Figure 4B). Post-hoc subgroup analysis demonstrated that augmented clinical benefit of avadomide was observed in patients with classifier-positive status, low ECOG PS, and normal LDH levels (supplemental Figure 5). Associations between these baseline variables were examined by univariate analysis of the classifier-positive status to ECOG PS (0-1 vs ≥ 2) and LDH level (normal vs elevated). Classifier status was not associated with LDH levels (χ2P = .878), nor was classifier status associated with ECOG PS status (Fisher exact test P = .5932) among the limited number of subjects with ECOG PS >2 (n = 3). Within the CC-122-ST-001 trial, a multivariate analysis could not be performed due to limited numbers of patients (< 5 patients) in some demographic or clinical subgroups.
Discussion
Cereblon modulators that degrade Aiolos and Ikaros have demonstrated clinical efficacy in B-cell malignancies, with lenalidomide showing preferential activity in ABC-DLBCL patients.26,53 Compared with lenalidomide, avadomide treatment results in a deeper and kinetically faster degradation of Aiolos and Ikaros, leading to COO-independent antiproliferative and tumor cell apoptotic activity in preclinical models.48,54,55 These observations, in addition to the potent immunomodulatory activity of avadomide, served as the basis for the current study.37 In this phase 1 trial in patients with R/R DLBCL including transformed lymphoma, avadomide monotherapy was well tolerated, with no unexpected safety concerns, consistent with previous findings in other advanced malignancies including solid tumors.45 Based on results of this study, the recommended phase 2 dose was determined to be 3 mg avadomide formulated capsule, administered on a 5/7-day schedule.
The current study provides preliminary data that avadomide is an active antilymphoma drug with acceptable tolerability. The most common AE (all grade and grade 3/4) observed with avadomide was neutropenia, which results from Ikaros degradation in myeloid cells, leading to a maturation arrest at a promyelocyte stage.56 The introduction of an intermittent 5/7-day dosing schedule improved tolerability and reduced the frequency and severity of AEs while maintaining the clinical activity of avadomide. We recently reported that lenalidomide-mediated degradation of Ikaros in an in vitro model of myeloid differentiation results in a reversible arrest in neutrophil maturation, which is released upon removal of the drug, without any loss of cell viability.52 Using a similar preclinical model, we demonstrated that treatment of human bone marrow precursor cells with avadomide on a novel 5/7-day schedule allowed Ikaros to be resynthesized during the 2-day drug holiday, enabling mature neutrophils to repopulate after a transient arrest.57
Our data demonstrate the potent immunomodulatory activity of avadomide, including positive effects on T-cell activation and a broad expansion of T-cell populations, as defined by an increase in richness of the peripheral T-cell repertoire. A diverse TCR repertoire and low clonal index of T-cell populations in tumors has been associated with prolonged survival in DLBCL patients.58 In addition, the results of this study demonstrate decreased proliferation of malignant B cells within the tumor, with concomitant increased trafficking of immune cells, such as macrophages, to the tumor microenvironment.
Avadomide demonstrated broad activity across DLBCL COO status, as observed preclinically in both in vitro and in vivo models.37 Interestingly, patients whose pretreatment tumors were positive for a novel gene expression classifier had significantly prolonged mPFS and higher ORR compared with classifier-negative patients, supporting the classifier as a strategy to enrich for R/R DLBCL patients likely to respond to avadomide. Classifier-negative R/R DLBCL patients who received avadomide monotherapy showed clinical responses comparable to those reported in patients treated with various salvage chemotherapy regimens and/or stem cell transplantation.12 As we describe in a companion paper,46 the classifier segregates patients with de novo DLBCL, based on the presence of high vs low immune cell infiltration within the tumor. The correlation between high immune infiltrate in tumors and improved outcome on avadomide described in this study, although preliminary, is consistent with the mechanisms of action of avadomide and its observed pharmacodynamic activities, such as T-cell activation and increased trafficking of macrophages to the DLBCL tumor. An understanding of the interplay between avadomide effects and the tumor microenvironment, as well as the biology associated with defined cell types within classifier-positive tumors, can inform patient selection strategies and combination strategies. Rational combinations that may have potential in current and future clinical trials include checkpoint blockade of the CD47/SIRPα signaling axis, CD20-targeting monoclonal antibodies such as rituximab or obinutuzumab (NCT02031419 and NCT02417285), the CD19-targeted chimeric antigen receptor T cell lisocabtagene maraleucel (NCT03310619), and R-CHOP chemoimmunotherapy (NCT03283202).
For data sharing, e-mail the corresponding author at ccarpio@vhio.net.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the coinvestigators on the CC-122-ST-001 study and all patients, families, and caregivers who participated. Medical writing was provided by Dorothy Fallows, and editorial assistance was provided by Bio Connections, LLC.
This study was supported by the Celgene Corporation, A Bristol-Myers Squibb Company.
Authorship
Contribution: Together with Bristol-Myers Squibb authors (M.P., A.K.G., P.R.H., X. Wei, K.H., and M.W.B.T.), C.C. and V.R. were responsible for study design and C.C., M.P., A.K.G., P.R.H., X. Wei, M.W., S. Couto, S. Carrancio, F.S., A.W., X. Wei, M.W.B.T., A.R., and V.R. contributed to data interpretation and analysis; and C.C., R.B., L.Y., J.-M.S., G.S., R.C., A.P., M.G., D.R., C.P., J.A.L-M., A. Santoro, A. Salar, S.D., A.M., G.V., E.V.d.N., M.P., T.J.B., A.K.G., P.R.H., X. Wang, S. Couto, S. Carrancio, F.S., A.W., X. Wei, and V.R. reviewed patient records and contributed to data collection. Celgene, A Bristol-Myers Squibb Company, was involved in study design, compilation of data, and statistical analysis. All authors had full access to all of the data, carefully reviewed the manuscript, and approved the final version. The corresponding author, C.C., had final responsibility to submit for publication.
Conflict-of-interest disclosure: C.C. received travel grants from Takeda, Janssen, Roche, and Celgene, A Bristol-Myers Squibb Company. L.Y. reports research funding from Janssen and Roche, and served on the advisory board for Abbvie, Gilead, Roche, and Janssen. J.-M.S. received travel grants from Roche and honoraria from Roche, Gilead, Janssen, Celgene, A Bristol-Myers Squibb Company, Kern-Pharma and Servier and served on the advisory board of Roche, Gilead, Janssen, Bristol-Myers Squibb, Kern-Pharma, and Celltrion. G.S. received research funding from Roche and Celgene, A Bristol-Myers Squibb Company, and served on the advisory board for Novartis. R.C. participated in a speaker’s bureau for Roche, Janssen, and Celgene, A Bristol-Myers Squibb Company; received honoraria from Roche, Janssen, and Celgene, A Bristol-Myers Squibb Company; served on the advisory board for Janssen, Celgene, A Bristol-Myers Squibb Company, and Servier; and received travel grants from Roche, Janssen, Celgene, A Bristol-Myers Squibb Company, AbbVie, and Pfizer. A.P. participated in a speaker’s bureau for Roche; received patents or royalties from EDO-Mundipharma; received honoraria from Roche, MSD, Bristol-Myers Squibb, and Servier; served on the advisory board of Servier, Roche, Bristol-Myers Squibb, and MSD; and received travel grants from Roche and Takeda. D.R. served on the advisory board of Boehringer Ingelheim and Eli Lilly, received travel grants from Asana, and received research funding from Celgene, A Bristol-Myers Squibb Company. C.P. participated in a speaker’s bureau for Roche and Janssen, served on the advisory board of Bristol-Myers Squibb and Kyowa Kirin, and received travel grants from Roche. J.A.L.-M. participated in a speaker’s bureau for Bristol-Myers Squibb, Roche, and MSD; received patents or royalties and reports equity ownership from PharmaMar; received travel grants from Bristol Myers-Squibb, Roche, and MSD; and received research funding from Iovance, Adaptimmune, Novartis, Roche, Merck, Pfizer, MSD, and Celgene, A Bristol-Myers Squibb Company. A. Santoro participated in a speaker’s bureau for Takeda, Roche, AbbVie, Amgen, Celgene, A Bristol-Myers Squibb Company, AstraZeneca, Eli Lilly, Sandoz, and Novartis and served on the advisory board for Bristol-Myers Squibb, Servier, Gilead, Pfizer, Eisai, Arqule, Bayer, and MSD. A. Salar participated in a speaker’s bureau for Roche and Janssen; received travel grants from Roche; served on the advisory board of Celgene, A Bristol-Myers Squibb Company, Roche, Janssen, Gilead; and reports research funding from Roche and Gilead. S.D. reports research funding from Novartis. A.M. received honoraria from Celgene, A Bristol-Myers Squibb Company, Roche, Janssen, and Servier; served on the advisory board of Roche, Celgene, A Bristol-Myers Squibb Company, and MorPhosys; provided expert testimony on behalf of Gilead; received travel grants from Roche, Celgene, A Bristol-Myers Squibb Company, Janssen, Servier, and Mundipharma; and reports research funding from Celgene, A Bristol-Myers Squibb Company, Janssen, Teva, and Mundipharma. A.W. received patents and royalties from BC Cancer Agency, received travel grants from Dava Oncology, served on the advisory board for m-panels and Guidepoint global, and reports research funding from Celgene, A Bristol-Myers Squibb Company. T.J.B. has equity ownership with Amgen. V.R. received honoraria from Infinity Pharmaceuticals, Bristol-Myers Squibb, Eisai, PharmaMar, and Gilead Sciences; served on the advisory board for Infinity Pharmaceuticals, Bristol-Myers Squibb, PharmaMar, Gilead Sciences, NanoString Technologies, Incyte, MSD, Roche, Genentech, and Epizyme; provided expert testimony on behalf of Servier, reports research funding from arGEN-X BVBA; and received travel grants from Roche and Bristol-Myers Squibb. F.S. has equity ownership with Bristol-Myers Squibb. S. Couto had equity ownership with Celgne, A Bristol-Myers Squibb Company. M.G., S. Carrancio, X. Wei, K.H., M.W.B.T., A.R., M.W., T.J.B., P.R.H., A.K.G., and M.P. are employees of Bristol-Myers Squibb and have equity ownership with Bristol-Myers Squibb. R.B., G.V., E.V.d.N., and X. Wang declare no competing financial interests.
Correspondence: Cecilia Carpio, University Hospital Vall d’Hebron, Passeig de la Vall d’Hebron, 119-129, 08035, Barcelona, Spain; e-mail: ccarpio@vhio.net.


![Best percentage reduction in target lesion burden over baseline in all response-evaluable patients. Dotted line indicates 50% reduction, consistent with a response (per investigator assessment, according to revised 2007 International Working Group [IWG] criteria). Blue, DLBCL de novo; red, transformed lymphoma.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/13/10.1182_blood.2019002395/7/m_bloodbld2019002395f3.png?Expires=1768443307&Signature=NrIF4FufK6gTQDWnQ~m9~2h5PODQjJ5HJwL1N5OZHkoHnZPG4ujh6jvH6iTnOiGomxfeM1M5DOQ1PfOVp~-EymEP2diH4HwoQAWQCh-thrxMr3cdFfSitjjuSyKOku54PNH05p9vwGXq8cByXJBdwk1qbiU5WQT-KCi1nJPb45PnpYT6-7TQoH2gTYfkncYcbWxs8QzZEyV5TFE3GUJRBtYTtZf96qwkFHkBuKJiwtgunXnjQ~Oy8mIXkLAquguckD~9NtI9syV12KEd32Ov1p5E-QFK9zQgg~3W7VQFmlvkkoml9ICy5grl9dXogYaO14yTS5Df5GrCdJdRzwaghw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)